Seroxat: Some of the adverse drug reactions listed below may decrease in intensity and frequency with continued treatment and do not generally lead to cessation of therapy. Adverse drug reactions are listed below by system organ class and frequency. Frequencies are defined as: very common (≥1/10), common (≥1/100, <1/10), uncommon (≥1/1,000, <1/100), rare (≥1/10,000, <1/1,000), very rare (<1/10,000), including isolated reports.
Blood & lymphatic system disorders: Uncommon: abnormal bleeding, predominantly of the skin and mucous membranes (including ecchymosis and gynaecological bleeding).
Very rare: thrombocytopenia.
Immune system disorders: Very rare: severe and potentially fatal allergic reactions (including anaphylactoid reactions and angioedema).
Endocrine disorders: Very rare: syndrome of inappropriate anti-diuretic hormone secretion (SIADH).
Metabolism & nutrition disorders: Common: increases in cholesterol levels, decreased appetite.
Uncommon: altered glycaemic control has been reported in diabetic patients (see Precautions).
Rare: hyponatraemia.
Hyponatraemia has been reported predominantly in elderly patients and is sometimes due to syndrome of inappropriate anti-diuretic hormone secretion (SIADH).
Psychiatric disorders: Common: somnolence, insomnia, agitation, abnormal dreams (including nightmares).
Uncommon: confusion, hallucinations.
Rare: manic reactions, anxiety, depersonalisation, panic attacks, akathisia (see Precautions).
Frequency not known: suicidal ideation, suicidal behaviour, aggression.
Cases of suicidal ideation and suicidal behaviour have been reported during paroxetine therapy or early after treatment discontinuation (see Precautions).
Cases of aggression were observed in post marketing experience.
These symptoms may also be due to the underlying disease.
Nervous system disorders: Common: dizziness, tremor, headache, concentration impaired.
Uncommon: extrapyramidal disorders.
Rare: convulsions, restless legs syndrome (RLS).
Very rare: serotonin syndrome (symptoms may include agitation, confusion, diaphoresis, hallucinations, hyperreflexia, myoclonus, shivering, tachycardia and tremor).
Reports of extrapyramidal disorders including oro-facial dystonia have been received in patients sometimes with underlying movement disorders or who were using neuroleptic medication.
Eye disorders: Common: blurred vision.
Uncommon: mydriasis (see Precautions).
Very rare: acute glaucoma.
Ear and labyrinth disorders: Frequency not known: tinnitus.
Cardiac disorders: Uncommon: sinus tachycardia.
Rare: bradycardia.
Vascular disorders: Uncommon: transient increases or decreases in blood pressure, postural hypotension.
Transient increases or decreases of blood pressure have been reported following treatment with paroxetine, usually in patients with pre-existing hypertension or anxiety.
Respiratory, thoracic and mediastinal disorders: Common: yawning.
Gastrointestinal disorders: Very common: nausea.
Common: constipation, diarrhoea, vomiting, dry mouth.
Very rare: gastrointestinal bleeding.
Hepato-biliary disorders: Rare: elevation of hepatic enzymes.
Very rare: hepatic events (such as hepatitis, sometimes associated with jaundice and/or liver failure).
Elevation of hepatic enzymes has been reported. Post-marketing reports of hepatic events (such as hepatitis, sometimes associated with jaundice and/or liver failure) have also been received very rarely. Discontinuation of paroxetine should be considered if there is prolonged elevation of liver function test results.
Skin & subcutaneous tissue disorders: Common: sweating.
Uncommon: skin rashes, pruritus.
Very rare: severe cutaneous adverse reactions (including erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis), urticaria, photosensitivity reactions.
Renal & urinary disorders: Uncommon: urinary retention, urinary incontinence.
Reproductive system & breast disorders: Very common: sexual dysfunction.
Rare: hyperprolactinaemia/galactorrhoea, menstrual disorders (including merorrhagia, metrorrhagia, amenorrhoea, menstruation delayed and menstruation irregular).
Very rare: priapism.
Musculoskeletal & connective tissue disorders: Rare: arthralgia, myalgia.
Epidemiological studies, mainly conducted in patients 50 years of age and older, show an increased risk of bone fractures in patients receiving SSRIs and TCAs. The mechanism leading to this risk is unknown.
General disorders & administration site conditions: Common: asthenia, body weight gain.
Very rare: peripheral oedema.
Withdrawal symptoms seen on discontinuation of paroxetine treatment: Common: Dizziness, sensory disturbances, sleep disturbances, anxiety, headache.
Uncommon: Agitation, nausea, tremor, confusion, sweating, emotional instability, visual disturbances, palpitations, diarrhoea, irritability.
Discontinuation of paroxetine (particularly when abrupt) commonly leads to withdrawal symptoms. Dizziness, sensory disturbances (including paraesthesia, electric shock sensations and tinnitus), sleep disturbances (including intense dreams), agitation or anxiety, nausea, tremor, confusion, sweating, headache, diarrhoea, palpitations, emotional instability, irritability and visual disturbances have been reported.
Generally these events are mild to moderate and are self-limiting; however, in some patients they may be severe and/or prolonged. It is therefore advised that when paroxetine treatment is no longer required, gradual discontinuation by dose tapering be carried out (see Dosage & Administration and Precautions).
Adverse Events from Paediatric Clinical Trials: The following adverse events were observed: Increased suicidal related behaviours (including suicide attempts and suicidal thoughts), self-harm behaviours and increased hostility. Suicidal thoughts and suicide attempts were mainly observed in clinical trials of adolescents with MDD. Increased hostility occurred particularly in children with obsessive compulsive disorder, and especially in younger children less than 12 years of age.
Additional events that were seen are: decreased appetite, tremor, sweating, hyperkinesia, agitation, emotional lability (including crying and mood fluctuations), bleeding related adverse events, predominantly of the skin and mucous membranes.
Events seen after discontinuation/tapering of paroxetine are: emotional lability (including crying, mood fluctuations, self-harm, suicidal thoughts and attempted suicide), nervousness, dizziness, nausea and abdominal pain (see Precaution).
See Pharmacology under Actions for more information on paediatric clinical trials.
Seroxat CR: Adverse Drug Reaction Overview: Commonly Observed Adverse Events: Depression: The most commonly observed adverse events associated with the use of SEROXAT CR in a pool of two trials (incidence of 5.0% or greater and incidence for SEROXAT CR at least twice that for placebo, derived from Table 8 below) were: abnormal ejaculation, abnormal vision, constipation, decreased libido, diarrhea, dizziness, female genital disorders, nausea, somnolence, sweating, trauma, tremor, and yawning.
Using the same criteria, the adverse events associated with the use of SEROXAT CR in a study of elderly patients with depression were: abnormal ejaculation, constipation, decreased appetite, dry mouth, impotence, infection, libido decreased, sweating, and tremor.
Panic Disorder: In the pool of panic disorder studies, the adverse events meeting these criteria were: abnormal ejaculation, somnolence, impotence, libido decreased, tremor, sweating, and female genital disorders (generally anorgasmia or difficulty achieving orgasm).
Social Anxiety Disorder: The most commonly observed adverse events associated with the use of SEROXAT CR (incidence of 5.0% or greater and incidence for SEROXAT CR at least twice that for placebo, derived from Table 11 as follows) in the social phobia (social anxiety disorder) study were nausea, asthenia, abnormal ejaculation, sweating, somnolence, impotence, insomnia, and libido decreased.
Premenstrual Dysphoric Disorder: The most commonly observed adverse events associated with the use of SEROXAT CR, either during continuous dosing or luteal phase dosing (incidence of 5.0% or greater and incidence for SEROXAT CR at least twice that for placebo, derived from Table 12 as follows) were: nausea, asthenia, libido decreased, somnolence, insomnia, female genital disorders, sweating, dizziness, diarrhea and constipation.
In the luteal phase dosing PMDD trial, which employed dosing of 12.5 mg/day or 25 mg/day of SEROXAT CR limited to the 2 weeks prior to the onset of menses over 3 consecutive menstrual cycles, adverse events were evaluated during the first 14 days of each off-drug phase. When the 3 off-drug phases were combined, the following adverse events were reported at an incidence of 2% or greater for SEROXAT CR and were at least twice the rate of that reported for placebo: Infection (5.3% versus 2.5%), depression (2.8% versus 0.8%), insomnia (2.4% versus 0.8%), sinusitis (2.4% versus 0%), and asthenia (2.0% versus 0.8%).
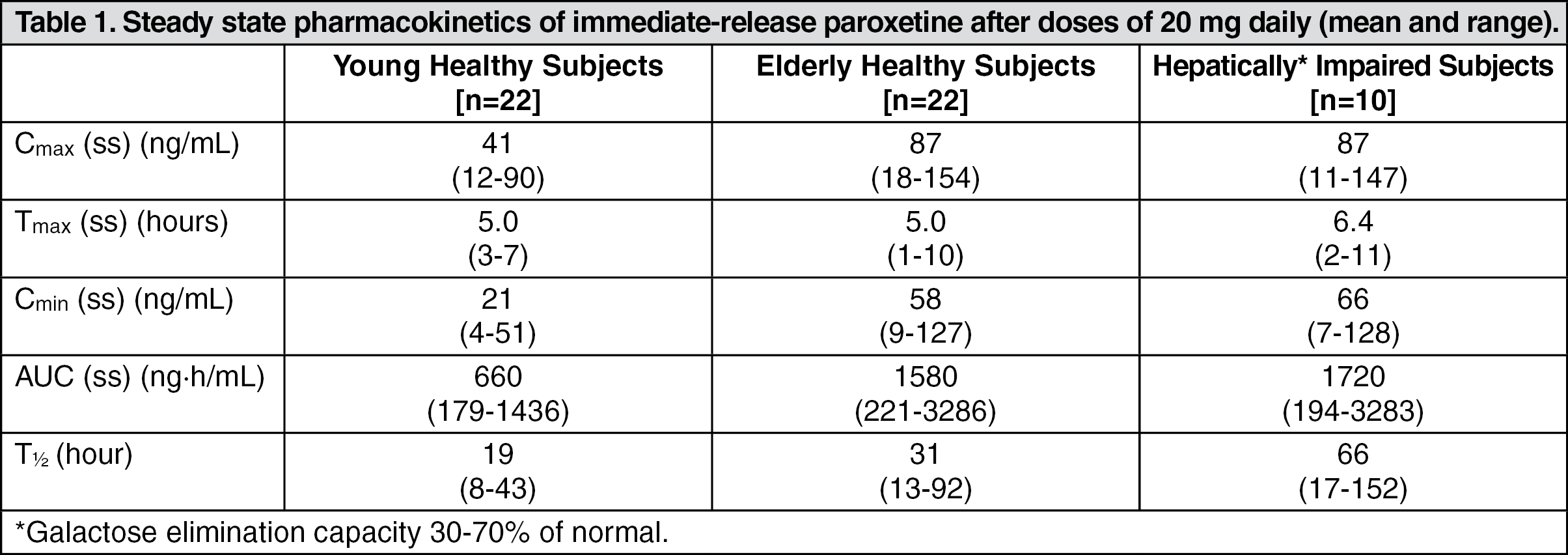
Adverse Events Leading to Discontinuation of Treatment: The information included under the "Adverse Events Leading to Discontinuation of Treatment" subsection of Adverse Reactions is based on data from seven short-term placebo-controlled clinical trials. Three of these studies were conducted in patients with depression, three studies were done in patients with panic disorder, and one study was conducted in patients with social anxiety disorder. Two of the studies in depression, which enrolled patients in the age range 18 to 65 years, are pooled. Information from a third study of depression, which focused on elderly patients (ages 60 to 88), is presented separately as is the information from the panic disorder studies and the information from the social anxiety disorder study. Information on additional adverse events associated with SEROXAT CR and the immediate-release formulation of paroxetine hydrochloride is included in a separate subsection.
Depression: Ten percent (21/212) of SEROXAT CR patients discontinued treatment due to an adverse event in a pool of two studies of patients with depression. The most common events (≥ 1%) associated with discontinuation and considered to be drug related (i.e., those events associated with dropout at a rate approximately twice or greater for SEROXAT CR compared to placebo) included the following: (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In a placebo-controlled study of elderly patients with depression, 13% (13/104) of SEROXAT CR patients discontinued due to an adverse event. Events meeting the above criteria included the following: (See Table 4.)
Click on icon to see table/diagram/image
Panic Disorder: Eleven percent (50/444) of SEROXAT CR patients in panic disorder studies discontinued treatment due to an adverse event. Events meeting the above criteria included the following: (See Table 5.)
Click on icon to see table/diagram/image
Social Anxiety Disorder: Three percent (5/186) of patients treated with SEROXAT CR in the social anxiety disorder study discontinued treatment due to an adverse event. Events meeting the above criteria included the following: (See Table 6.)
Click on icon to see table/diagram/image
Premenstrual Dysphoric Disorder: Thirteen percent (88/681) of patients treated with SEROXAT CR in PMDD studies of continuous dosing discontinued treatment due to an adverse event. Nine percent (34/366) of patients treated with SEROXAT CR in PMDD studies of luteal phase dosing discontinued treatment due to an adverse event.
The most common events (> 1%) associated with discontinuation and considered to be drug related (i.e., those events associated with dropout at a rate approximately twice or greater for SEROXAT CR compared to placebo) included the following: (See Table 7.)
Click on icon to see table/diagram/image
Adverse Events following Discontinuation of Treatment (or Dose Reduction): Clinical Trials: Adverse events while discontinuing therapy with SEROXAT CR were not systematically evaluated in most clinical trials; however, in one placebo-controlled clinical trial in social anxiety disorder involving 370 patients (186 on SEROXAT CR and 184 on placebo), utilizing daily doses of SEROXAT CR up to 37.5 mg/day, spontaneously reported adverse events while discontinuing therapy with SEROXAT CR were evaluated. Patients receiving 37.5 mg/day underwent an incremental decrease in the daily dose by 12.5 mg/day to a dose of 25 mg/day for 1 week before treatment was stopped. For patients receiving 25 mg/day or 12.5 mg/day, treatment was stopped without an incremental decrease in dose. With this regimen, the following adverse events were reported at an incidence of 2% or greater for SEROXAT CR and were at least twice that reported for placebo: dizziness (13.9 versus 2.2%), insomnia (4.4 versus 2.2%), paresthesia (4.4 versus 0%) vertigo (3.3 versus 0%), and additional symptoms described by the investigator as associated with tapering or discontinuing SEROXAT CR including electric shock sensations (5.6 versus 0.6%). These events were reported as serious in 1.7% (3/180) of patients who discontinued therapy with SEROXAT CR. The following adverse events have been reported at an incidence of 2% or greater for immediate-release paroxetine and were at least twice that reported for placebo: abnormal dreams (2.3 vs 0.5%), paresthesias (2.0 vs 0.4%), and dizziness (7.1 vs 1.5%). The majority of these events were mild to moderate, self-limiting and did not require medical intervention. These adverse events were noted in GAD and PTSD clinical trials employing a taper phase regimen for discontinuation of treatment. This regimen involved an incremental decrease in the daily dose by 10 mg/day at weekly intervals. When a daily dose of 20 mg/day was reached, patients were continued on this dose for 1 week before treatment was stopped.
Post-Marketing: There have been spontaneous reports of adverse events upon the discontinuation of paroxetine and SEROXAT CR (particularly when abrupt), including but not limited to the following: dizziness, sensory disturbances (including paresthesias, electric shock sensations and tinnitus), agitation/restlessness, anxiety, nausea, tremor, confusion, diarrhea, vomiting, sweating, headache, and sleep disturbances (abnormal dreams). Generally these symptoms are mild to moderate; however, in some patients they may be severe in intensity. They usually occur within the first few days of discontinuing treatment, but there have been very rare reports of such symptoms in patients who have inadvertently missed a dose. Generally these symptoms are self-limiting and usually resolve within 2 weeks, though in some individuals they may be prolonged (2-3 months or more). Symptoms associated with discontinuation have been reported for other selective serotonin reuptake inhibitors.
Patients should be monitored for these or any other symptoms when discontinuing treatment. If intolerable symptoms occur following a decrease in the dose or upon discontinuation of treatment, dose titration should be managed on the basis of the patient's clinical response (see Precautions and Dosage & Administration).
Clinical Trial Adverse Drug Reactions: Because clinical trials are conducted under very specific conditions the adverse reaction rates observed in the clinical trials may not reflect the rates observed in practice and should not be compared to the rates in the clinical trials of another drug. Adverse drug reaction information from clinical trials is useful for identifying drug-related adverse events and for approximating rates.
Incidence in Controlled Clinical Trials: Adults: Table 8 enumerates adverse events that occurred at an incidence of 1% or more among SEROXAT CR-treated patients, aged 18 to 65, who participated in two short-term (12-week) placebo-controlled trials in depression in which patients were dosed in a range of 25 to 62.5 mg/day. Table 9 enumerates adverse events reported at an incidence of 5% or greater among elderly SEROXAT CR-treated patients (ages 60-88) who participated in a short-term (12-week) placebo-controlled trial in depression in which patients were dosed in a range of 12.5 to 50 mg/day. Table 10 enumerates adverse events reported at an incidence of 1% or greater among SEROXAT CR treated patients (ages 19-72) who participated in short-term (10-week) placebo-controlled trials in panic disorder in which patients were dosed in a range of 12.5 to 75 mg/day. Table 11 enumerates adverse events reported at an incidence of 1% or greater among adult patients treated with SEROXAT CR who participated in a short-term (12-week) double-blind, placebo-controlled trial in Table 12 social anxiety disorder in which patients were dosed at a range of 12.5 to 37.5 mg/day enumerates adverse events that occurred at an incidence of 1% or more among SEROXAT CR treated patients who participated in three 12-week placebo-controlled trials in PMDD in which patients were dosed at 12.5 mg/day or 25 mg/day and in one 12-week placebo-controlled trial in which patients were dosed for 2 weeks prior to the onset of menses (luteal phase dosing) at 12.5 mg/day or 25 mg/day. Reported adverse events were classified using a standard COSTART-based dictionary terminology. The prescriber should be aware that these figures cannot be used to predict the incidence of side effects in the course of usual medical practice where patient characteristics and other factors differ from those which prevailed in the clinical trials. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigations involving different treatments, uses and investigators. The cited figures, however, do provide the prescribing physician with some basis for estimating the relative contribution of drug and nondrug factors to the side effect incidence rate in the population studied. (See Tables 8, 9, 10, 11, and 12.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Dose Dependency of Adverse Events: The following table shows results in PMDD trials of common adverse events, defined as events with an incidence of 1% with 25 mg of SEROXAT CR that was at least twice that with 12.5 mg of SEROXAT CR and with placebo. (See Table 13.)
Click on icon to see table/diagram/image
A comparison of adverse event rates in a fixed-dose study comparing immediate-release paroxetine with placebo in the treatment of depression revealed a clear dose dependency for some of the more common adverse events associated with the use of immediate-release paroxetine.
Male and Female Sexual Dysfunction with SSRIs: Although changes in sexual desire, sexual performance and sexual satisfaction often occur as manifestations of a psychiatric disorder, they may also be a consequence of pharmacologic treatment. In particular, some evidence suggests that selective serotonin reuptake inhibitors (SSRIs) can cause such untoward sexual experiences.
Reliable estimates of the incidence and severity of untoward experiences involving sexual desire, performance and satisfaction are difficult to obtain, however, in part because patients and physicians may be reluctant to discuss them. Accordingly, estimates of the incidence of untoward sexual experience and performance, cited in product labeling, are likely to underestimate their actual incidence.
The percentage of patients reporting symptoms of sexual dysfunction in the pool of two placebo-controlled trials in non-elderly patients with depression, in the pool of three placebo-controlled trials in patients with panic disorder, in the placebo-controlled trial in patients with social anxiety disorder, and in the luteal phase dosing and in the pool of 3 placebo-controlled trials in female patients with PMDD are as follows: See Table 14.
Click on icon to see table/diagram/image
There are no adequate, controlled studies examining sexual dysfunction with paroxetine treatment.
Paroxetine treatment has been associated with several cases of priapism. In those cases with a known outcome, patients recovered without sequelae.
While it is difficult to know the precise risk of sexual dysfunction associated with the use of SSRIs, physicians should routinely inquire about such possible side effects.
Laboratory Changes - Cholesterol: Clinically and statistically relevant increases in cholesterol levels have been noted in studies using paroxetine (see Precautions).
Of the patients in placebo-controlled clinical trials for whom baseline and on-treatment measurements were taken, total serum levels of cholesterol showed a mean increase of ~ 1.5 mg/dL in paroxetine-treated patients (n = 653), compared to a mean decrease of ~ 5.0 mg/dL in placebo-treated patients (n = 379). Increases from baseline of 45 mg/dL or greater were recorded in 6.6% of paroxetine-treated patients compared to 2.6% of placebo-treated patients.
Pediatrics: In placebo-controlled clinical trials conducted with pediatric patients aged 7 to 18 years with depression, OCD and Social Anxiety Disorder (involving 633 patients treated with paroxetine and 542 patients treated with placebo), the following adverse events were reported in at least 2% of pediatric patients treated with immediate-release paroxetine and occurred at a rate at least twice that for pediatric patients receiving placebo: emotional lability (including self-harm, suicidal thoughts, attempted suicide, crying, and mood fluctuations), hostility, (predominantly aggression, oppositional behaviour and anger) decreased appetite, tremor, sweating, hyperkinesia, and agitation.
In the pediatric clinical trials in depression, OCD and Social Anxiety: Disorder that included a taper phase regimen (307 patients aged 7 to 18 years treated with paroxetine and 291 patients treated with placebo), events reported upon discontinuation of treatment, which occurred in at least 2% of patients who received immediate-release paroxetine and which occurred at a rate at least twice that of placebo, were: emotional lability (including suicidal ideation, suicide attempt, mood changes, and tearfulness), nervousness, dizziness, nausea, and abdominal pain (see Precautions).
Other Events Observed During the Clinical Development of Paroxetine: The following adverse events were reported during the clinical development of SEROXAT CR tablets and/or the clinical development of the immediate-release formulation of paroxetine. Adverse events for which frequencies are provided below occurred in clinical trials with the controlled release formulation of paroxetine. During its premarketing assessment in depression, panic disorder, social anxiety disorder, and PMDD, multiple doses of SEROXAT CR were administered to 1627 patients in phase 3 double-blind, controlled, outpatient studies. Untoward events associated with this exposure were recorded by clinical investigators using terminology of their own choosing. Consequently, it is not possible to provide a meaningful estimate of the proportion of individuals experiencing adverse events without first grouping similar types of untoward events into a smaller number of standardized event categories.
In the tabulations that follow, reported adverse events were classified using a COSTART-based dictionary. The frequencies presented, therefore, represent the proportion of the 1627 patients exposed to SEROXAT CR controlled release who experienced an event of the type cited on at least one occasion while receiving SEROXAT CR. All reported events are included except those already listed in Tables 8, 9, 10, 11 or 12 and those events where a drug cause was remote. If the COSTART term for an event was so general as to be uninformative, it was deleted or, when possible, replaced with a more informative term. It is important to emphasize that although the events reported occurred during treatment with paroxetine, they were not necessarily caused by it.
Events are further categorized by body system and listed in order of decreasing frequency according to the following definitions: frequent adverse events are those occurring on one or more occasions in at least 1/100 patients (only those not already listed in the tabulated results from placebo-controlled trials appear in this listing); infrequent adverse events are those occurring in 1/100 to 1/1000 patients; rare events are those occurring in fewer than 1/1000 patients.
Adverse events for which frequencies are not provided occurred during the premarketing assessment of immediate-release paroxetine in phase 2 and 3 studies of depression, obsessive compulsive disorder, panic disorder, social anxiety disorder, generalized anxiety disorder, and posttraumatic stress disorder. The conditions and duration of exposure to immediate-release paroxetine varied greatly and included (in overlapping categories) open and double-blind studies, uncontrolled and controlled studies, inpatient and outpatient studies, and fixed-dose and titration studies. Only those events not previously listed for controlled release paroxetine are included. The extent to which these events may be associated with SEROXAT CR is unknown.
Events are listed alphabetically within the respective body system. Events of major clinical importance are also described in the Precautions section.
Body as a Whole: Infrequent were chills, face edema, fever, flu syndrome, malaise; rare were abscess, anticholinergic syndrome, hypothermia; also observed were adrenergic syndrome, neck rigidity, sepsis.
Immune System Disorders: Very rare were severe allergic reactions (including anaphylactoid reactions and angioedema).
Cardiovascular System: Infrequent were angina pectoris, bradycardia, bundle branch block, hematoma, hypertension, hypotension, palpitation, postural hypotension, supraventricular tachycardia, syncope; rare was bundle branch block; also observed were arrhythmia nodal, atrial fibrillation, cerebrovascular accident, congestive heart failure, low cardiac output, myocardial infarct, myocardial ischemia, pallor, phlebitis, pulmonary embolus, supraventricular extrasystoles, thrombophlebitis, thrombosis, vascular headache, ventricular extrasystoles.
Digestive System: Infrequent were bruxism, dysphagia, eructation, gastritis, gastroenteritis, gastroesophageal reflux, gingivitis, hemorrhoids, liver function tests abnormal, melena, pancreatitis, rectal hemorrhage, toothache, ulcerative stomatitis; rare were colitis, glossitis, gum hyperplasia, hepatosplenomegaly, increased salivation, intestinal obstruction, peptic ulcer, stomach ulcer, throat tightness; also observed were aphthous stomatitis, bloody diarrhea, bulimia, cardiospasm, cholelithiasis, duodenitis, enteritis, esophagitis, fecal impactions, fecal incontinence, gum hemorrhage, hematemesis, hepatitis, ileitis, ileus, jaundice, mouth ulceration, salivary gland enlargement, sialadenitis, stomatitis, tongue discoloration, tongue edema.
Endocrine System: Infrequent were, ovarian cyst, testes pain; rare were diabetes mellitus, hyperthyroidism; also observed were, goiter, hypothyroidism, thyroiditis.
Hemic and Lymphatic System: Infrequent were anemia, eosinophilia, hypochromic anemia, leukocytosis, leukopenia, lymphadenopathy, purpura; rare was thrombocytopenia; also observed were anisocytosis, basophilia, bleeding time increased, lymphedema, lymphocytosis, lymphopenia, microcytic anemia, monocytosis, normocytic anemia, thrombocythemia.
Metabolic and Nutritional Disorders: Frequent were increases in cholesterol levels. Infrequent were generalized edema, hyperglycemia, hyperkalemia, hypokalemia, peripheral edema, SGOT increased, SGPT increased, thirst; rare were billirubinemia, dehydration, hyperkalemia, obesity; also observed were alkaline phosphatase increased, BUN increased, creatinine phosphokinase increased, gamma globulins increased, gout, hypercalcemia, hyperphosphatemia, hypocalcemia, hypoglycemia, hyponatremia, ketosis, lactic dehydrogenase increased, non-protein nitrogen (NPN) increased.
Musculoskeletal System: Infrequent were arthritis, bursitis, tendonitis; rare were myasthenia, myopathy, myositis; also observed were generalized spasm, osteoporosis, tenosynovitis, tetany.
Nervous System: Frequent was depression; infrequent were amnesia, convulsion, depersonalization, dystonia, emotional lability, hallucinations, hyperkinesia, hypesthesia, hypokinesia, incoordination, libido increased, neuralgia, neuropathy, nystagmus, paralysis, vertigo; rare were ataxia, coma, diplopia, dyskinesia, hostility, paranoid reaction, torticollis, withdrawal syndrome; also observed were abnormal gait, akathisis, akinesia, aphasia, choreoathetosis, circumoral paresthesia, delirium, delusions, dysarthria, euphoria, extrapyramidal syndrome, fasciculations, grand mal convulsion, hyperalgesia, irritability, manic reaction, manic-depressive reaction, meningitis, myelitis, peripheral neuritis, psychosis, psychotic depression, reflexes decreased, reflexes increased, stupor, trismus.
Respiratory System: Frequent was pharyngitis; infrequent were asthma, dyspnea, epistaxis, laryngitis, pneumonia; rare was stridor; also observed were dysphonia, emphysema, hemoptysis, hiccups, hyperventilation, lung fibrosis, pulmonary edema, respiratory flu, sputum increased.
Skin and Appendages: Frequent was rash; infrequent were acne, alopecia, dry skin, eczema, pruritus, urticaria; rare were exfoliative dermatitis, furunculosis, pustular rash, seborrhea; also observed were angioedema, ecchymosis, erythema multiforme, erythema nodosum, hirsutism, maculopapular rash, skin discoloration, skin hypertrophy, skin ulcer, sweating decreased, vesiculobullous rash; very rare were severe cutaneous adverse reactions (including erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis).
Special Senses: Infrequent were conjunctivitis, earache, keratoconjunctivitis, mydriasis, photophobia, retinal hemorrhage, tinnitus; rare were blepharitis, visual field defect; also observed were amblyopia, anisocoria, blurred vision, cataract, conjunctival edema, corneal ulcer, deafness, exophthalmos, glaucoma, hyperacusis, night blindness, parosmia, ptosis, taste loss.
Urogenital System: Frequent was dysmennorhea*; infrequent were albuminuria, amenorrhea*, breast pain*, cystitis, dysuria, prostatitis*, urinary retention; rare were breast enlargement*, breast neoplasm*, female lactation, hematuria, kidney calculus, metrorrhagia, nephritis, nocturia, pregnancy and puerperal disorders*, salpingitis, urinary incontinence, uterine fibroids enlarged*; also observed were breast atrophy, ejaculatory disturbance, endometrial disorder, epididymitis, fibrocystic breast, leukorrhea, mastitis, oliguria, polyuria, pyuria, urethritis, urinary casts, urinary urgency, urolith, uterine spasm, vaginal hemorrhage.
*Based on the number of men and women as appropriate.
Post-Marketing Adverse Drug Reactions: Adverse events not listed above which have been reported since market introduction in patients taking immediate-release paroxetine hydrochloride include acute pancreatitis, hepatic events such as elevation of hepatic enzymes, and hepatitis, sometimes associated with jaundice, and/or liver failure (in very rare circumstances, with fatal outcomes), Guillain-Barre syndrome, priapism, thrombocytopenia, aggravated hypertension, syndrome of inappropriate ADH secretion, symptoms suggestive of hyperprolactinemia and galactorrhea, menstrual disorders (including menorrhagia, metrorrhagia and amenorrhea), blurred vision, extrapyramidal symptoms which have included akathisia, (characterized by an inner sense of restlessness and psychomotor agitation such as an inability to sit or stand still usually associated with subjective distress), bradykinesia, cogwheel rigidity, dystonia, hypertonia, oculogyric crisis which has been associated with concomitant use of pimozide, tremor and trismus, abnormal dreams (including nightmares), restless legs syndrome (RLS), vomiting, neuroleptic malignant syndrome-like events; serotonin syndrome (see Precautions), persistent pulmonary hypertension (PPHN; see also Precautions). There has been a case report of an elevated phenytoin level after 4 weeks of immediate-release paroxetine and phenytoin co-administration. There has been a case report of severe hypotension when immediate-release paroxetine was added to chronic metoprolol treatment. The causal relationship between immediate-release paroxetine and the emergence of these events has not been established.
There have been spontaneous reports of adverse events upon the discontinuation of SEROXAT CR and other selective serotonin reuptake inhibitors (particularly when abrupt) (see Precautions and Adverse Reactions).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image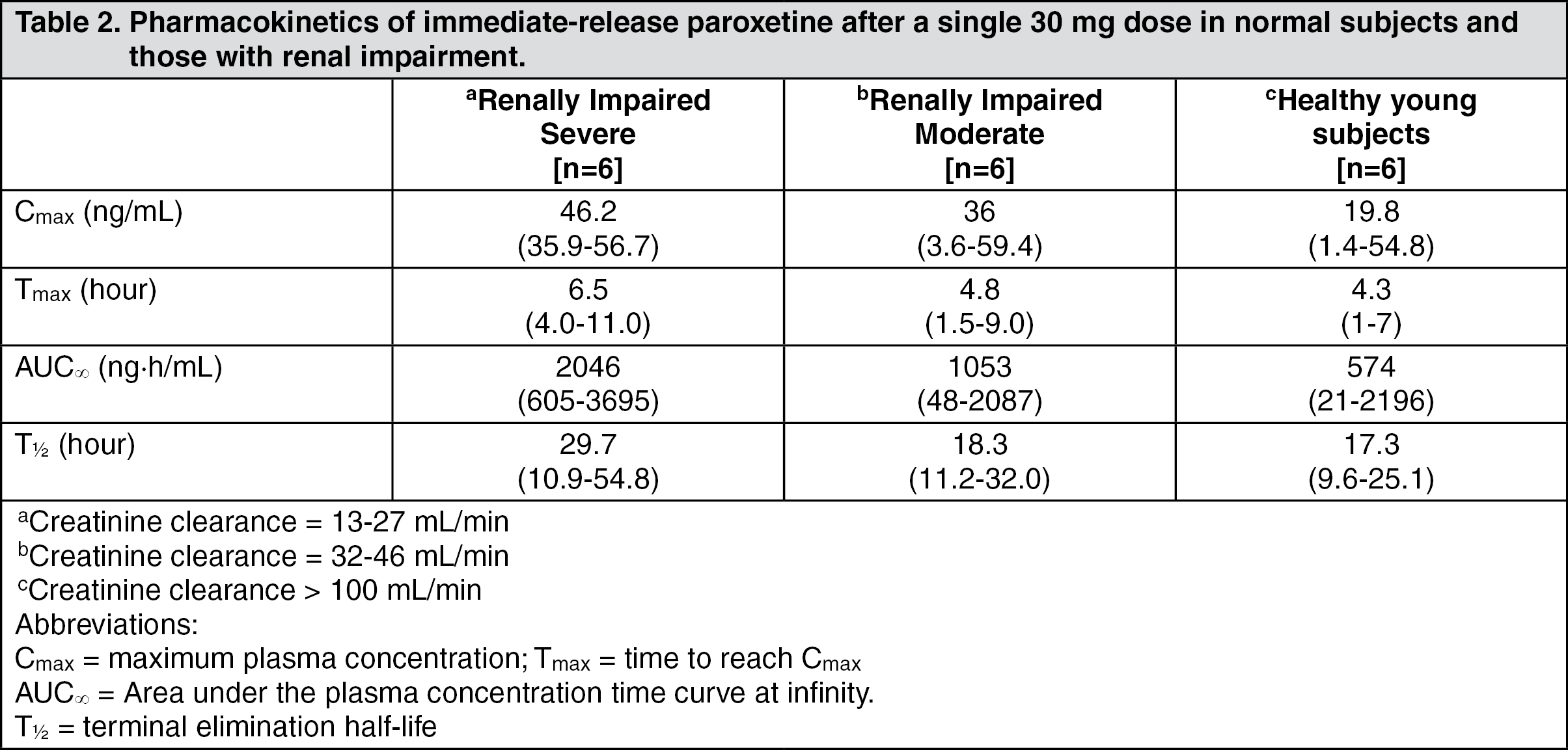 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image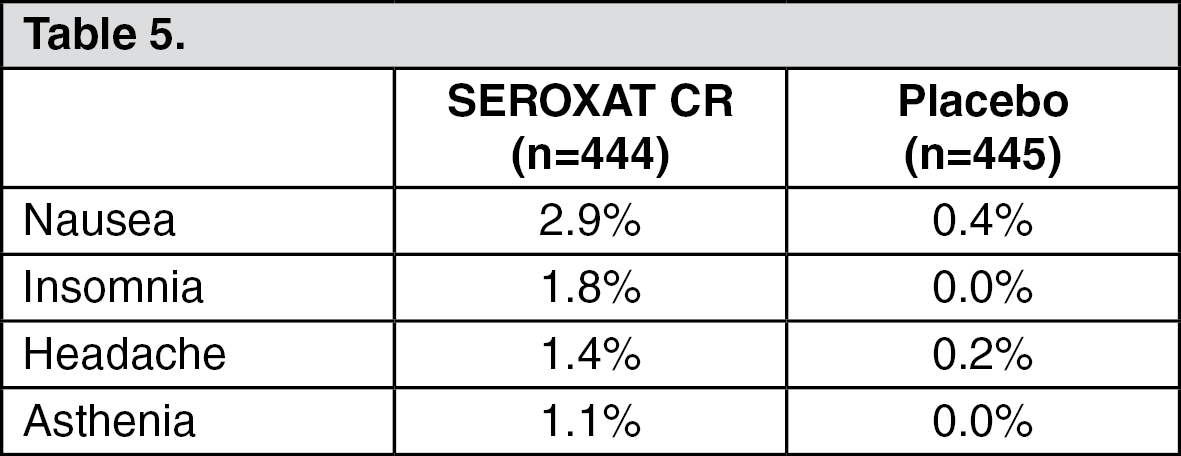 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image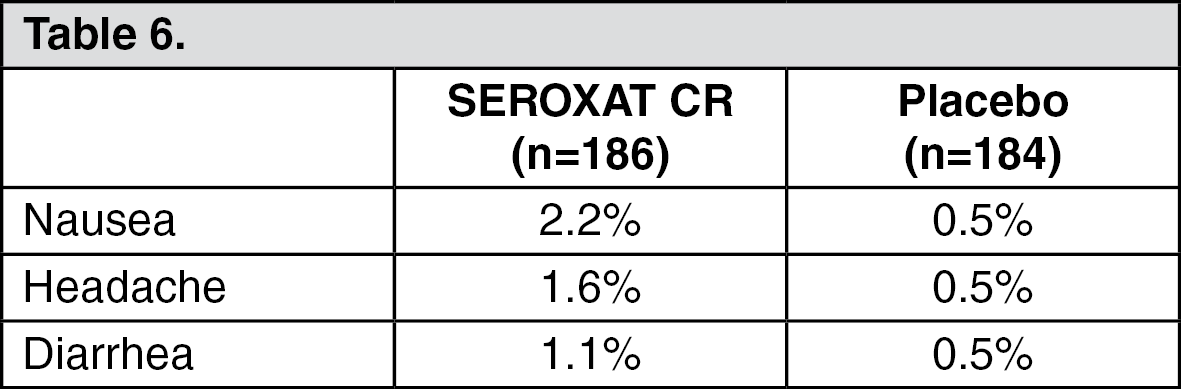 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image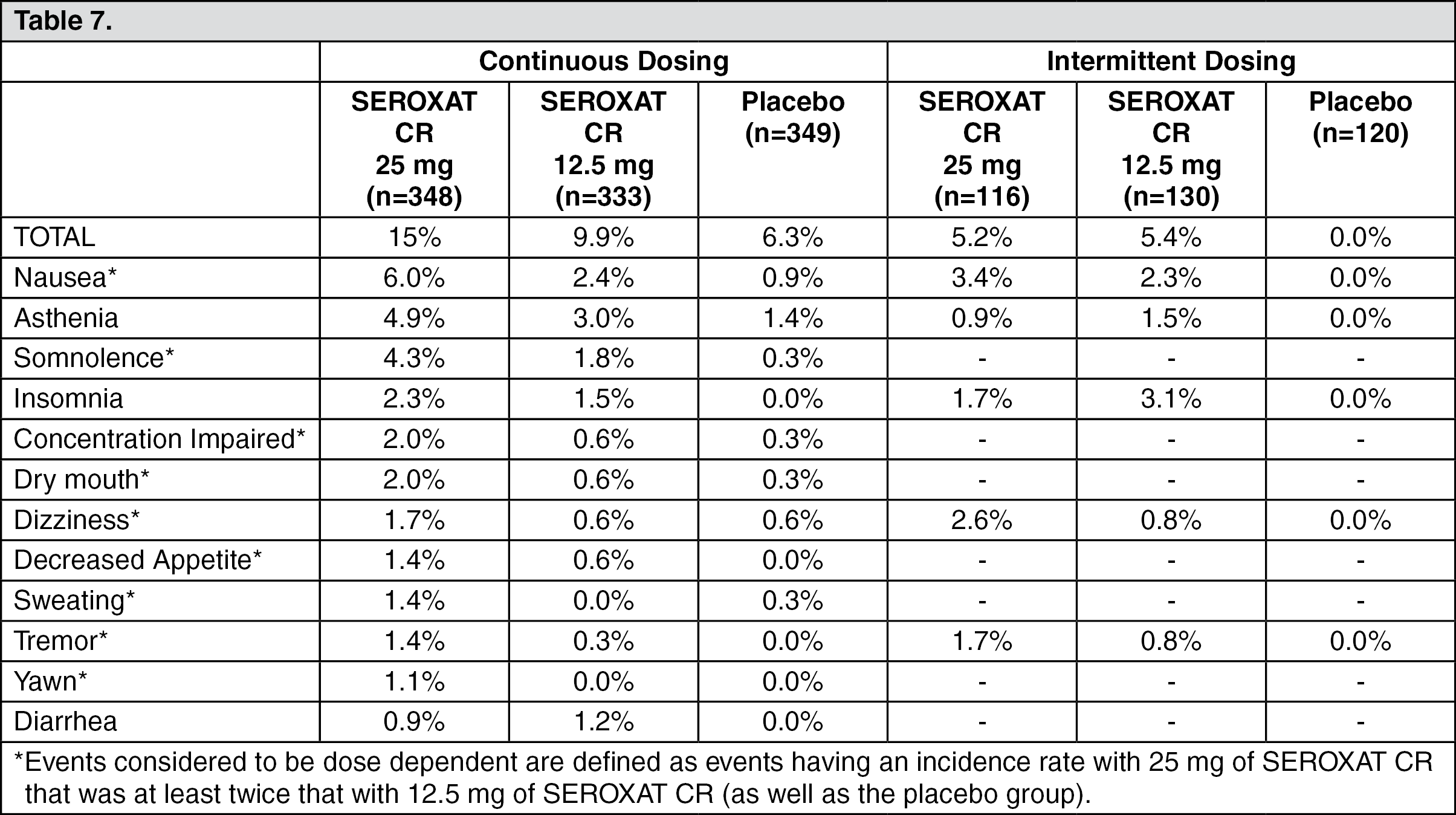 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image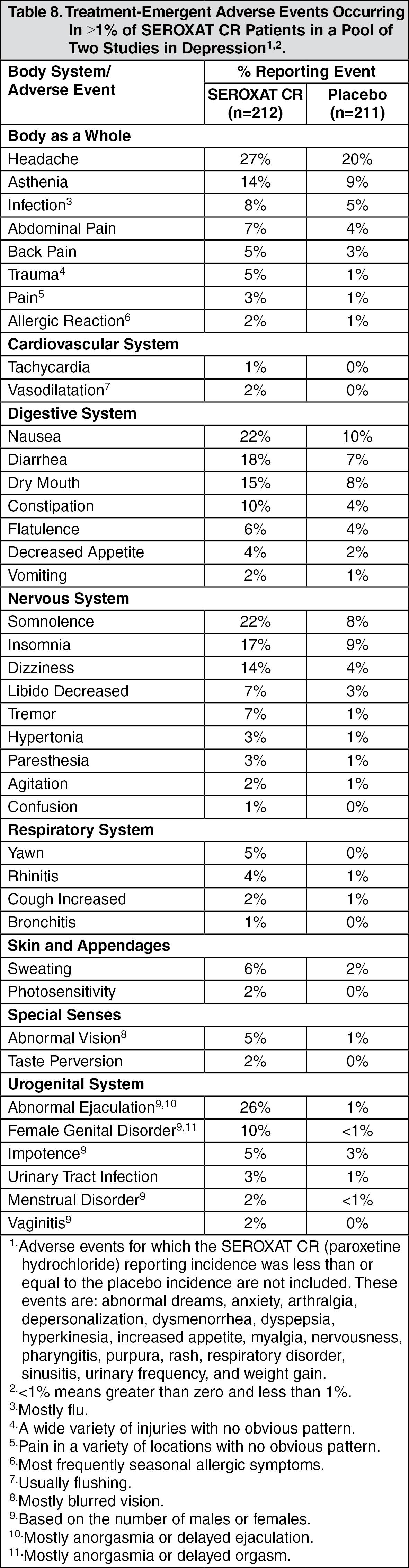 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image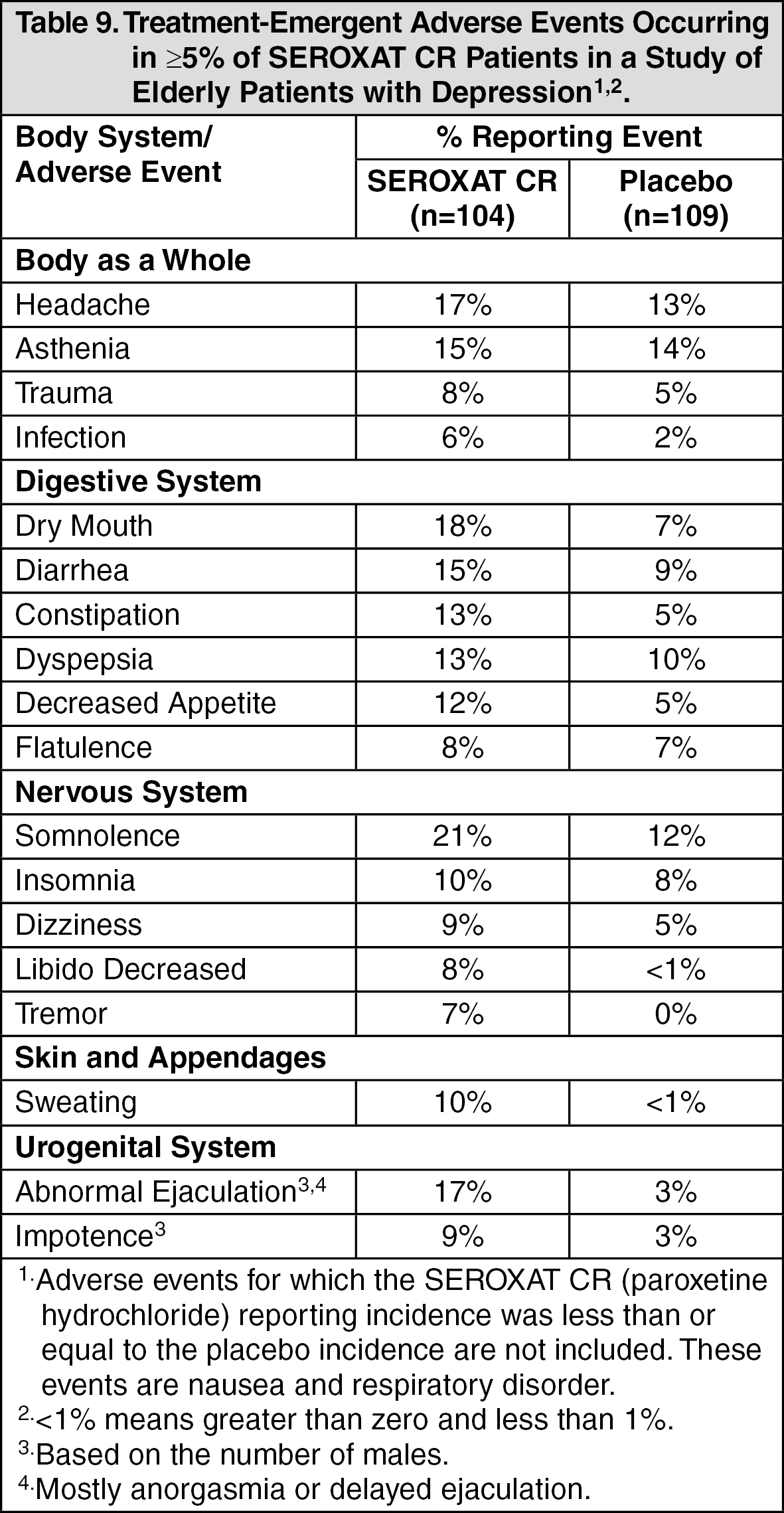 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image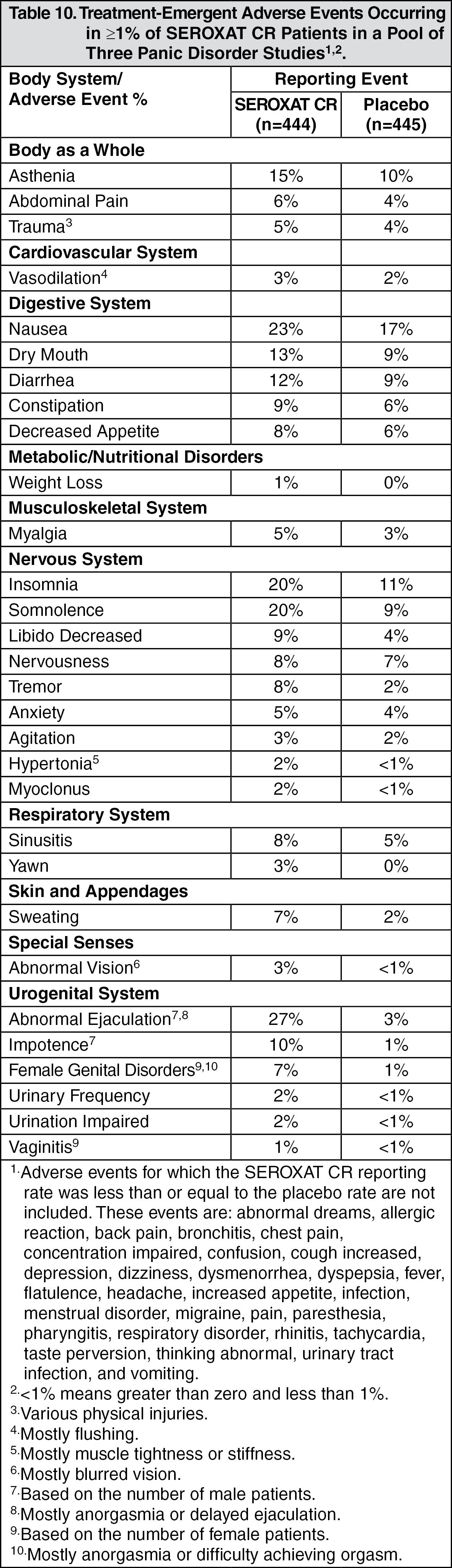 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image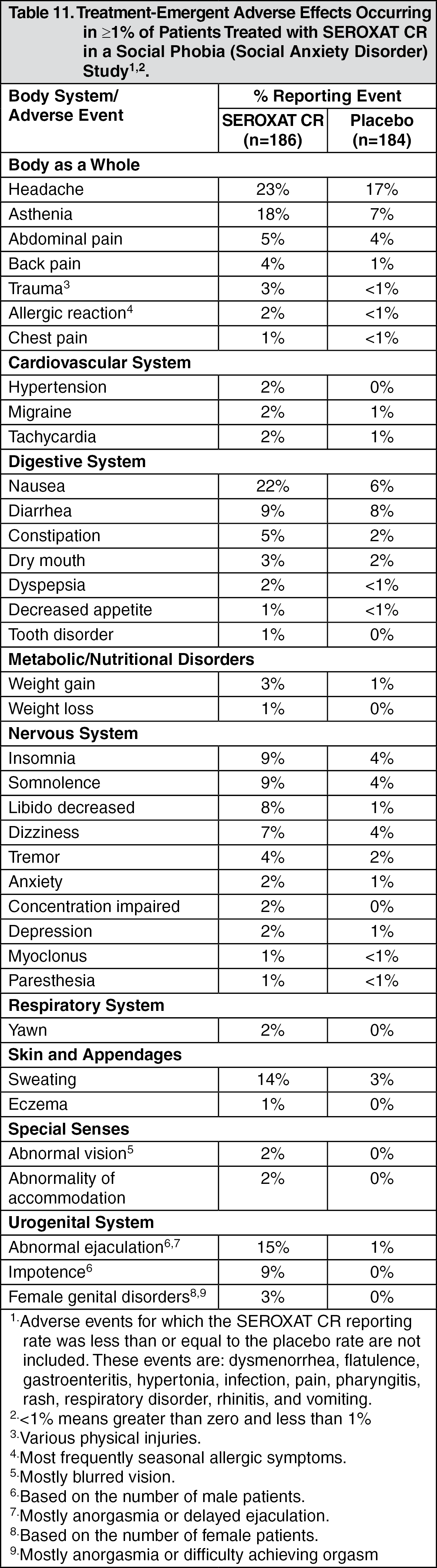 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image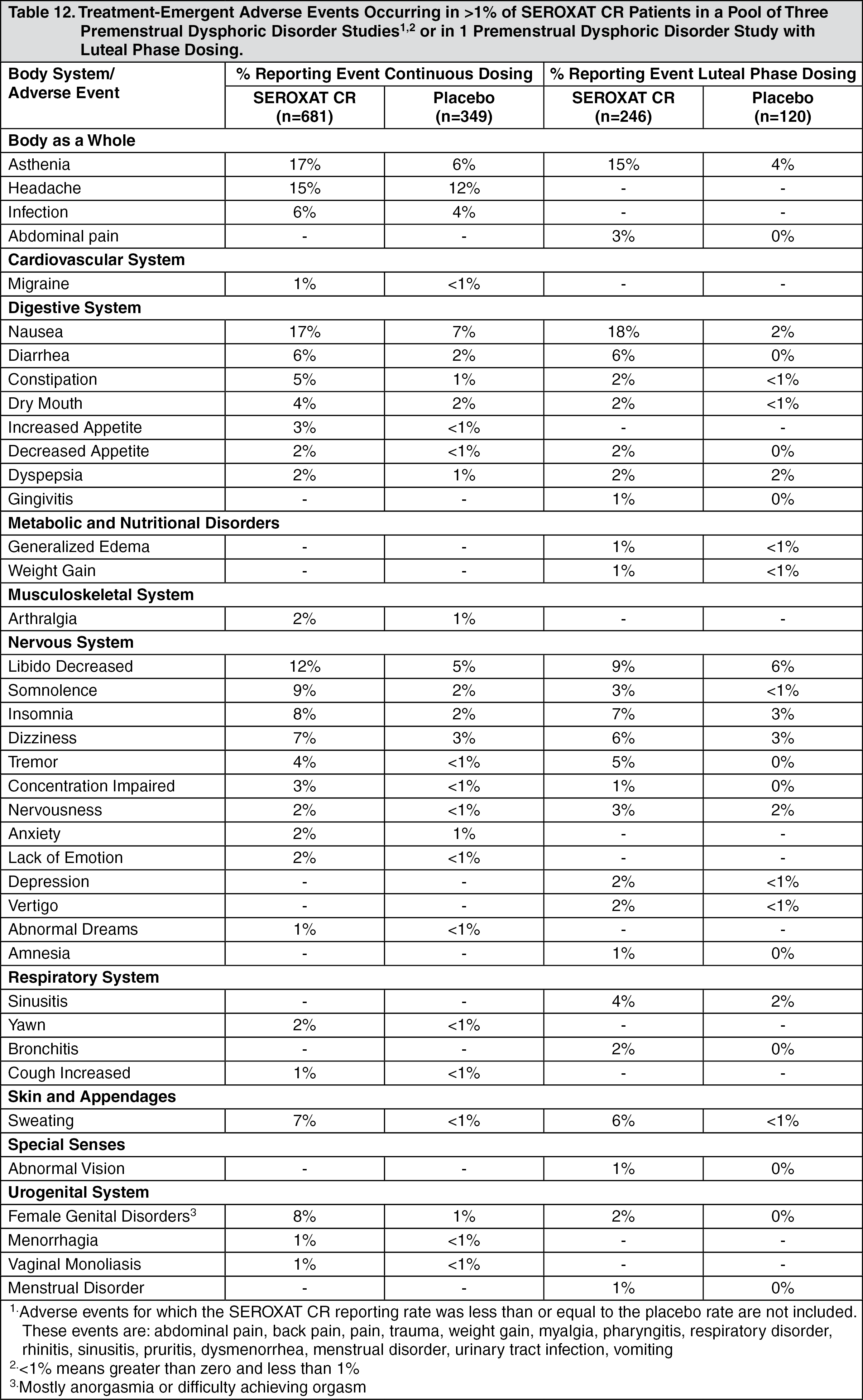 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image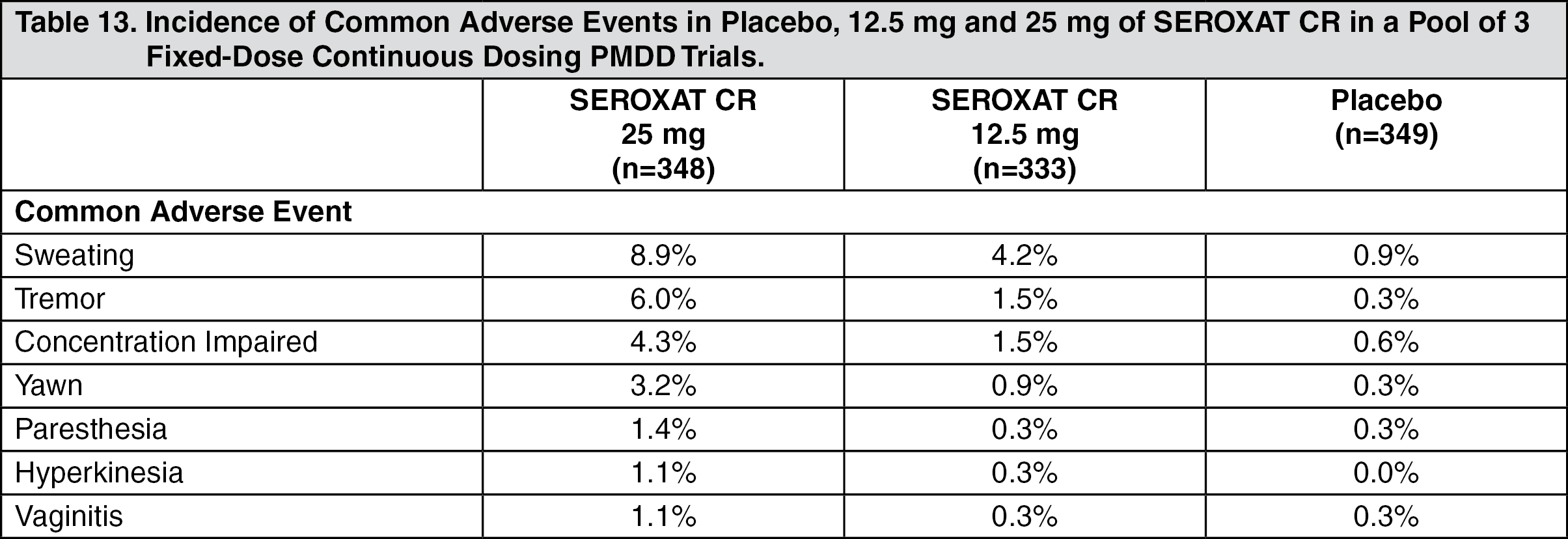 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image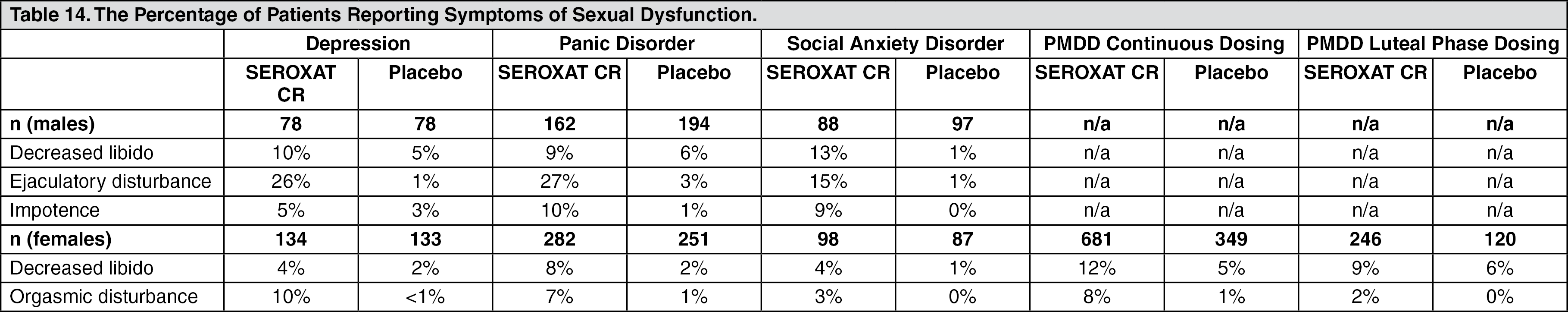 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image



 Sign Out
Sign Out
