Therapeutic Class: FOSAMAX PLUS contains alendronate sodium and cholecalciferol (vitamin D
3).
Alendronate Sodium: Alendronate sodium is a bisphosphonate that acts as a potent, specific inhibitor of osteoclast-mediated bone resorption. Bisphosphonates are synthetic analogs of pyrophosphate that bind to the hydroxyapatite found in bone.
Cholecalciferol: Cholecalciferol (vitamin D
3) is a secosterol that is the natural precursor of the calcium-regulating hormone calcitriol (1,25-dihydroxyvitamin D
3).
Pharmacology: Mechanism of Action: Alendronate Sodium: Animal studies have indicated the following mode of action. At the cellular level, alendronate shows preferential localization to sites of bone resorption specifically under osteoclasts. The osteoclasts adhere normally to the bone surface but lack the ruffled border that is indicative of active resorption. Alendronate does not interfere with osteoclast recruitment or attachment, but it does inhibit osteoclast activity. Studies in mice on the localization of radioactive [
3H] alendronate in bone showed about 10-fold higher uptake on osteoclast surfaces than on osteoblast surfaces. Bones examined 6 and 49 days after [
3H] alendronate administration in rats and mice, respectively, showed that normal bone was formed on top of the alendronate, which was incorporated inside the matrix, where it is no longer pharmacologically active. Thus, alendronate must be continuously administered to suppress osteoclasts on newly formed resorption surfaces. Histomorphometry in baboons and rats showed that alendronate treatment reduces bone turnover (i.e., the number of sites at which bone is remodeled). In addition, bone formation exceeds bone resorption at these remodeling sites, leading to progressive gains in bone mass.
Cholecalciferol: Vitamin D
3 is produced in the skin by photochemical conversion of 7-dehydrocholesterol to previtamin D
3 by ultraviolet light. This is followed by non-enzymatic isomerization to vitamin D
3. In the absence of adequate sunlight exposure, vitamin D
3 is an essential dietary nutrient. Vitamin D
3 in skin and dietary vitamin D
3 (absorbed into chylomicrons) is converted to 25-hydroxyvitamin D
3 in the liver. Conversion to the active calcium-mobilizing hormone 1,25-dihydroxyvitamin D
3 (calcitriol) in the kidney is stimulated by both parathyroid hormone and hypophosphatemia. The principal action of 1,25-dihydroxyvitamin D
3 is to increase intestinal absorption of both calcium and phosphate as well as regulate serum calcium, renal calcium and phosphate excretion, bone formation and bone resorption.
Vitamin D
3 is required for normal bone formation. Vitamin D insufficiency develops when both sunlight exposure and dietary intake are inadequate. Insufficiency is associated with negative calcium balance, bone loss, and increased risk of skeletal fracture. In severe cases, deficiency results in secondary hyperparathyroidism, hypophosphatemia, proximal muscle weakness and osteomalacia, further increasing the risk of falls and fractures in osteoporotic individuals. Supplemental vitamin D reduces these risks and their consequences.
Pharmacodynamics: Alendronate is a bisphosphonate that binds to bone hydroxyapatite and specifically inhibits the activity of osteoclasts, the bone-resorbing cells. Alendronate reduces bone resorption with no direct effect on bone formation, although the latter process is ultimately reduced because bone resorption and formation are coupled during bone turnover.
Osteoporosis in postmenopausal women: Osteoporosis is characterized by low bone mass that leads to an increased risk of fracture. It occurs in both males and females but is most common among women following the menopause, when bone turnover increases and the rate of bone resorption exceeds that of bone formation. These changes result in progressive bone loss and lead to osteoporosis in a significant proportion of women over age 50. Fractures, usually of the spine, hip, and wrist, are the common consequences. From age 50 to age 90, the risk of hip fracture in women increases 50-fold and the risk of vertebral fracture 15- to 30-fold. It is estimated that approximately 40% of 50-year-old women will sustain one or more osteoporosis-related fractures of the spine, hip, or wrist during their remaining lifetimes. Hip fractures, in particular, are associated with substantial morbidity, disability, and mortality.
Daily oral doses of alendronate (5, 20, and 40 mg for six weeks) in postmenopausal women produced biochemical changes indicative of dose-dependent inhibition of bone resorption, including decreases in urinary calcium and urinary markers of bone collagen degradation (such as hydroxyproline, deoxypyridinoline, and cross-linked N-telopeptides of type I collagen). These biochemical changes returned toward baseline values as early as three weeks following the discontinuation of therapy with alendronate and did not differ from placebo after 7 months despite the long retention of alendronate in the skeleton.
Long-term treatment of osteoporosis with FOSAMAX 10 mg/day (for up to five years) reduced urinary excretion of markers of bone resorption, deoxypyridinoline and cross-linked N-telopeptides of type l collagen, by approximately 50% and 70%, respectively, to reach levels similar to those seen in healthy premenopausal women. The decrease in the rate of bone resorption indicated by these markers was evident as early as one month and at three to six months reached a plateau that was maintained for the entire duration of treatment with FOSAMAX. In osteoporosis treatment studies, FOSAMAX 10 mg/day decreased the markers of bone formation, osteocalcin and bone specific alkaline phosphatase by approximately 50%, and total serum alkaline phosphatase by approximately 25 to 30%, to reach a plateau after 6 to 12 months. Similar reductions in the rate of bone turnover were observed in postmenopausal women during a one-year study with FOSAMAX once weekly 70 mg for the treatment of osteoporosis. These data indicate that the rate of bone turnover reached a new steady-state, despite the progressive increase in the total amount of alendronate deposited within bone.
As a result of inhibition of bone resorption, asymptomatic reductions in serum calcium and phosphate concentrations were also observed following treatment with FOSAMAX. In the long-term studies, reductions from baseline in serum calcium (approximately 2%) and phosphate (approximately 4 to 6%) were evident the first month after the initiation of FOSAMAX 10 mg. No further decreases in serum calcium were observed for the five-year duration of treatment, however, serum phosphate returned toward prestudy levels during years 3 through 5. In one-year studies with FOSAMAX once weekly 70 mg, similar reductions were observed at 6 and 12 months. The reduction in serum phosphate may reflect not only the positive bone mineral balance due to FOSAMAX but also a decrease in renal phosphate reabsorption.
Osteoporosis in men: Even though osteoporosis is less prevalent in men than in postmenopausal women, a significant proportion of osteoporotic fractures occur in men. The prevalence of vertebral deformities appears to be similar in men and women. Treatment of men with osteoporosis with FOSAMAX 10 mg/day for two years reduced urinary excretion of cross-linked N-telopeptides of type I collagen by approximately 60% and bone-specific alkaline phosphatase by approximately 40%. Similar reductions were observed in a one-year study in men with osteoporosis receiving FOSAMAX once weekly 70 mg.
Clinical Studies: Treatment of osteoporosis: FOSAMAX PLUS studies: The effect of FOSAMAX PLUS (alendronate 70 mg/vitamin D
3 2800 IU) on vitamin D status was demonstrated in a 15-week, double-blind, multinational study of 717 osteoporotic postmenopausal women and men (serum 25-hydroxyvitamin D at baseline: mean, 22.2 ng/mL [56 nmol/L]; range, 9-90 ng/mL [22.5-225 nmol/L]). Patients received FOSAMAX PLUS (70 mg/2800 IU) (n=350 women, 10 men) or FOSAMAX (alendronate) 70 mg (n=332 women, 25 men) once a week; additional vitamin D supplements were prohibited. The percentage of patients with serum 25-hydroxyvitamin D ≥ 15 ng/mL (37.5 nmol/L) was significantly higher with FOSAMAX PLUS (70 mg/2800 IU) vs. alendronate only (89% vs. 68%, respectively). The percentage of patients with serum 25-hydroxyvitamin D ≥ 9 ng/mL (22.5 nmol/L) was significantly higher with FOSAMAX PLUS (70 mg/2800 IU) vs. alendronate only (99% vs 87%, respectively). There were no differences in mean serum calcium, phosphate, or 24-hour urine calcium between treatment groups.
The effect of FOSAMAX PLUS (alendronate 70 mg/vitamin D
3 2800 IU) with an additional 2800 IU vitamin D
3 for a total of 5600 IU once weekly was demonstrated in a 24-week, extension study that enrolled 652 osteoporotic post-menopausal women and men. Patients in the Vitamin D
3 2800 group received FOSAMAX PLUS (70 mg/2800 IU) (n=305 women, 21 men) and those in the Vitamin D
3 5600 group received FOSAMAX PLUS (70 mg/2800 IU) with an additional 2800 IU vitamin D
3 (n=314 women, 12 men) once a week; additional vitamin D supplements were allowed. After 24-weeks of treatment, the mean serum 25-hydroxyvitamin D levels were significantly higher in the Vitamin D
3 5600 group (27.9 ng/ml [70 nmol/l]) than in the Vitamin D
3 2800 group (25.6 ng/ml [64 nmol/l]). The percentage of patients with serum 25-hydroxyvitamin D ≥ 15 ng/mL (37.5 nmol/L) was higher with the Vitamin D
3 5600 group vs. the Vitamin D
3 2800 group (96.9% vs. 94.4%, respectively). The percentage of patients with serum 25-hydroxyvitamin D ≥ 9 ng/mL (22.5 nmol/L) was higher with the Vitamin D
3 5600 group vs. the Vitamin D
3 2800 group (100% vs. 99.7%, respectively) through the 24-week extension. There were no differences in mean serum calcium, phosphate, or 24-hour urine calcium between treatment groups. The percentage of patients with hypercalciuria at the end of the 24-week extension was not statistically different between treatment groups.
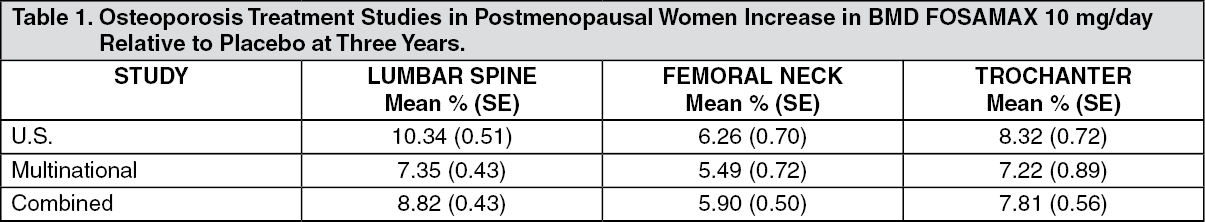
FOSAMAX (alendronate sodium) studies: Postmenopausal women: Effect on bone mineral density: The efficacy of FOSAMAX 10 mg once daily in postmenopausal women with osteoporosis was demonstrated in four double-blind, placebo-controlled clinical studies of two or three years' duration. These included two large three-year, multicenter studies of virtually identical design, one performed in the United States (U.S.) and the other in 15 different countries (Multinational), which enrolled 478 and 516 patients, respectively. The following table shows the mean increases in bone mineral density (BMD) of the lumbar spine, femoral neck, and trochanter in patients receiving FOSAMAX 10 mg/day relative to placebo-treated patients at three years for each of these studies. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In the combined studies, after three years, BMD of the lumbar spine, femoral neck and trochanter in placebo-treated patients decreased significantly by between 0.65% and 1.16%. Highly significant increases, relative both to baseline and placebo, were seen at each measurement site in each study in patients who received FOSAMAX 10 mg/day. Total body BMD also increased significantly in both studies, indicating that the increases in bone mass of the spine and hip did not occur at the expense of other skeletal sites. Increases in BMD were evident as early as three months and continued throughout the entire three years of treatment. In the two-year extension of these studies, treatment with FOSAMAX 10 mg/day resulted in continued increases in BMD at the lumbar spine and trochanter (absolute additional increases between years 3 and 5: lumbar spine, 0.94%; trochanter, 0.88%). BMD at the femoral neck, forearm and total body were maintained. Thus, FOSAMAX reverses the progression of osteoporosis. FOSAMAX was similarly effective regardless of age, race, baseline rate of bone turnover, renal function, and use with a wide range of common medications.
In a separate study, FOSAMAX 10 mg/day for two years induced highly significant increases in BMD of the spine, femoral neck, trochanter, and total body relative to either intranasal salmon calcitonin 100 IU/day or placebo.
In patients with postmenopausal osteoporosis treated with FOSAMAX 10 mg/day for one or two years the effects of treatment withdrawal were assessed. Following discontinuation, bone turnover gradually returned toward pretreatment levels, and BMD no longer increased although accelerated bone loss was not observed. These data indicate that treatment with FOSAMAX must be continuous to produce progressive increases in bone mass.
The therapeutic equivalence of FOSAMAX once weekly 70 mg (n = 519) and FOSAMAX 10 mg daily (n = 370) was demonstrated in a one-year, double-blind, multicenter study of postmenopausal women with osteoporosis. The mean increases from baseline in lumbar spine BMD at one year were 5.1% (4.8, 5.4%; 95% CI) in the 70 mg once-weekly group and 5.4% (5.0, 5.8%; 95% CI) in the 10 mg daily group. The two treatment groups were also similar with regard to BMD increases at other skeletal sites. These data support the expectation that FOSAMAX once weekly 70 mg will have effects to reduce the incidence of fractures similar to those of daily treatment (see as follows).
Effect on fracture incidence: To assess the effects of FOSAMAX on vertebral fracture incidence, the U.S. and Multinational studies were combined in an analysis that compared placebo to the pooled dosage groups of FOSAMAX (5 or 10 mg for three years or 20 mg for two years followed by 5 mg for one year). There was a statistically significant and clinically meaningful 48% reduction in the proportion of patients treated with FOSAMAX experiencing one or more vertebral fractures relative to those treated with placebo (3.2% vs. 6.2%). An even greater reduction in the total number of vertebral fractures (4.2 vs. 11.3 per 100 patients) was also observed. Furthermore, of patients who sustained any vertebral fracture, those treated with FOSAMAX experienced less height loss (5.9 mm vs. 23.3 mm) due to a reduction in both the number and severity of fractures.
Additionally, analysis of the data pooled across doses of ≥ 2.5 mg from five placebo-controlled studies of two or three years' duration including the U.S. and Multinational studies (FOSAMAX, n = 1012; placebo, n = 590) revealed a significant 29% reduction in non-vertebral fracture incidence (FOSAMAX, 9.0% vs. placebo, 12.6%). Like the effect on vertebral fracture incidence, these results of alendronate treatment are consistent with the observed increases in bone mass.
The Fracture Intervention Trial (FIT) consisted of two studies in postmenopausal women: the Three-Year Study of patients who had at least one baseline vertebral (compression) fracture and the Four-Year Study of patients with low bone mass but without a baseline vertebral fracture.
Fracture Intervention Trial: Three-Year Study (patients with at least one baseline vertebral fracture): This randomized, double-blind, placebo-controlled, 2027-patient study (FOSAMAX, n=1022; placebo, n=1005) demonstrated that treatment with FOSAMAX resulted in statistically significant and clinically meaningful reductions in fracture incidence at three years as shown in the table as follows. Proportionately similar reductions of hip and wrist fractures were seen in the five pooled osteoporosis treatment studies (see as previously mentioned). (See Table 2.)
Click on icon to see table/diagram/image
Furthermore, in this population of patients with baseline vertebral fracture, treatment with FOSAMAX significantly reduced the incidence of hospitalizations resulting from any cause (25.0% vs. 30.7%, a 20% reduction). This difference appears to be related, at least in part, to the reduction in fracture incidence.
Fracture Intervention Trial: Four-Year Study (patients with low bone mass but without a baseline vertebral fracture): This randomized, double-blind, placebo-controlled, 4432-patient study (FOSAMAX, n=2214; placebo, n=2218) further demonstrated the reduction in fracture incidence were due to FOSAMAX. The intent of the study was to recruit women with osteoporosis, i.e. with a baseline femoral neck BMD at least two standard deviations below the mean for young adult women. However, due to subsequent revisions to the normative values for femoral neck BMD, 31% of patients were found not to meet this entry criterion and thus this study included both osteoporotic and non-osteoporotic women. The results are shown in the table as follows for the patients with osteoporosis. (See Table 3.)
Click on icon to see table/diagram/image
In all patients (including those without osteoporosis), the reductions in fracture incidence were: ≥ 1 painful fracture, 14% (p = 0.072); ≥1 vertebral fracture, 44% (p = 0.001); ≥1 painful vertebral fracture, 34% (p = 0.178), and hip fracture, 21% (p = 0.44). The incidence of wrist fracture in all patients was FOSAMAX, 3.7%; placebo, 3.2% (not significant).
Combined FIT Studies: The reductions in fracture incidence for the combined Three- and Four-Year Studies of FIT are shown as follows. (See Table 4.)
Click on icon to see table/diagram/image
Consistency of fracture results: The reductions in the incidence of vertebral fractures (FOSAMAX versus placebo) in the Three- and Four-Year Studies of FIT were consistent with that in the combined U.S. and Multinational (U.S./Mult) treatment studies (see as previously mentioned), in which 80% of the women did not have a vertebral fracture at baseline. During these studies, treatment with FOSAMAX reduced the proportion of women experiencing at least one new vertebral fracture by approximately 50% (Three-Year FIT: 47% reduction, p < 0.001; Four-Year FIT: 44% reduction, p = 0.001; U.S./Mult: 48% reduction, p = 0.034). In addition, FOSAMAX reduced the proportion of women experiencing multiple (two or more) new vertebral fractures by approximately 90% in the U.S./Mult. and Three-Year FIT studies (p<0.001). Thus, FOSAMAX reduces the incidence of vertebral fractures whether or not patients have experienced a previous vertebral fracture.
Overall, these results demonstrate the consistent efficacy of FOSAMAX to reduce the incidence of fractures, including those of the spine and hip, which are the sites of osteoporotic fracture associated with the greatest morbidity.
Bone histology: Bone histology in 270 postmenopausal patients with osteoporosis treated with FOSAMAX at doses ranging from 1 to 20 mg/day for one, two or three years revealed normal mineralization and structure, as well as the expected decrease in bone turnover relative to placebo. These data, together with the normal bone histology and increased bone strength observed in rats and baboons exposed to long-term alendronate treatment, indicate that bone formed during therapy with FOSAMAX is of normal quality.
Men: The efficacy of FOSAMAX in men with osteoporosis was demonstrated in two clinical studies.
A two-year, double-blind, placebo-controlled, multicenter study of FOSAMAX 10 mg once daily enrolled a total of 241 men between the ages of 31 and 87 (mean, 63). At two years, the mean increases relative to placebo in BMD in men receiving FOSAMAX 10 mg/day were: lumbar spine, 5.3%; femoral neck, 2.6%; trochanter, 3.1%; and total body, 1.6% (all p≤0.001). Consistent with much larger studies in postmenopausal women, in these men, FOSAMAX 10 mg/day reduced the incidence of new vertebral fracture (assessed by quantitative radiography) relative to placebo (0.8% vs. 7.1%, respectively; p=0.017) and, correspondingly, also reduced height loss (-0.6 vs. -2.4 mm, respectively; p=0.022).
A one-year, double-blind, placebo-controlled, multicenter study of FOSAMAX once weekly 70 mg enrolled a total of 167 men between the ages of 38 and 91 (mean 66). At one year, the mean increases in BMD relative to placebo were significant at the following sites: lumbar spine, 2.8% (p ≤ 0.001); femoral neck, 1.9% (p = 0.007); trochanter, 2.0% (p ≤ 0.001); and total body, 1.2% (p = 0.018). These increases in BMD were similar to those seen at one year in the 10 mg once-daily study.
In both studies FOSAMAX was effective regardless of age, gonadal function or baseline BMD (femoral neck and lumbar spine).
Concomitant use with estrogen/hormone replacement therapy (HRT): The effects on BMD of treatment with FOSAMAX 10 mg once daily and conjugated estrogen (0.625 mg/day) either alone or in combination were assessed in a two-year, double-blind, placebo-controlled study of hysterectomized postmenopausal osteoporotic women (n=425). At two years, the increases in lumbar spine BMD from baseline were significantly greater with the combination (8.3%) than with either estrogen or FOSAMAX alone (both 6.0%).
The effects on BMD when FOSAMAX was added to stable doses (for at least one year) of HRT (estrogen ± progestin) were assessed in a one-year, double-blind, placebo-controlled study in postmenopausal osteoporotic women (n=428). The addition of FOSAMAX 10 mg once daily to HRT produced, at one year, significantly greater increases in lumbar spine BMD (3.7%) vs. HRT alone (1.1%).
In these studies, significant increases or favorable trends in BMD for combined therapy compared with HRT alone were seen at the total hip, femoral neck, and trochanter. No significant effect was seen for total body BMD.
Pharmacokinetics: Absorption: Alendronate Sodium: Relative to an intravenous (IV) reference dose, the mean oral bioavailability of alendronate in women was 0.64% for doses ranging from 5 to 70 mg when administered after an overnight fast and two hours before a standardized breakfast. Oral bioavailability in men (0.6%) was similar to that in women.
The alendronate in the FOSAMAX PLUS (70 mg/2800 IU) and FOSAMAX PLUS (70 mg/5600 IU) tablets and the FOSAMAX 70 mg tablet is bioequivalent.
Bioavailability was decreased similarly (by approximately 40%) whether alendronate was administered one or one-half hour before a standardized breakfast. In osteoporosis studies, FOSAMAX was effective when administered at least 30 minutes before the first food or beverage of the day.
Bioavailability was negligible whether alendronate was administered with or up to two hours after a standardized breakfast. Concomitant administration of alendronate with coffee or orange juice reduced bioavailability by approximately 60%.
In healthy subjects, oral prednisone (20 mg three times daily for five days) did not produce a clinically meaningful change in the oral bioavailability of alendronate (a mean increase ranging from 20 to 44%).
Cholecalciferol: Following administration of FOSAMAX PLUS (70 mg/2800 IU) after an overnight fast and two hours before a standard meal, the mean area under the serum-concentration-time curve (AUC
0-120 hrs) for vitamin D
3 (unadjusted for endogenous vitamin D
3 levels) was 296.4 ng-hr/mL. The mean maximal serum concentration (C
max) of vitamin D
3 was 5.9 ng/mL, and the median time to maximal serum concentration (T
max) was 12 hrs. Following administration of FOSAMAX PLUS (70 mg/5600 IU) after an overnight fast and two hours before a meal, the mean area under the serum-concentration-time curve (AUC
0-80 hrs) for vitamin D
3 (unadjusted for endogenous vitamin D
3 levels) was 490.2 ng-hr/ml. The mean maximal serum concentration (C
max) of vitamin D
3 was 12.2 ng/ml and the median time to maximal serum concentration (T
max) was 10.6 hours. The bioavailability of the vitamin D
3 in FOSAMAX PLUS (70 mg/2800 IU) and FOSAMAX PLUS (70 mg/5600 IU) is similar to an equal dose of vitamin D
3 administered alone.
Distribution: Alendronate Sodium: Studies in rats show that alendronate transiently distributes to soft tissues following 1 mg/kg IV administration but is then rapidly redistributed to bone or excreted in the urine. The mean steady state volume of distribution, exclusive of bone, is at least 28 L in humans. Concentrations of drug in plasma following therapeutic oral doses are too low for analytical detection (less than 5 ng/mL). Protein binding in human plasma is approximately 78%.
Cholecalciferol: Following absorption, vitamin D
3 enters the blood as part of chylomicrons. Vitamin D
3 is rapidly distributed mostly to the liver where it undergoes metabolism to 25-hydroxyvitamin D
3, the major storage form. Lesser amounts are distributed to adipose and muscle tissue and stored as vitamin D
3 at these sites for later release into the circulation. Circulating vitamin D
3 is bound to vitamin D-binding protein.
Metabolism: Alendronate Sodium: There is no evidence that alendronate is metabolized in animals or humans.
Cholecalciferol: Vitamin D
3 is rapidly metabolized by hydroxylation in the liver to 25-hydroxyvitamin D
3, and subsequently metabolized in the kidney to 1,25-dihydroxyvitamin D
3, which represents the biologically active form. Further hydroxylation occurs prior to elimination. A small percentage of vitamin D
3 undergoes glucuronidation prior to elimination.
Elimination: Alendronate Sodium: Following a single IV dose of [
14C] alendronate, approximately 50% of the radioactivity was excreted in the urine within 72 hours and little or no radioactivity was recovered in the feces.
Following a single 10 mg IV dose, the renal clearance of alendronate was 71 mL/min, and systemic clearance did not exceed 200 mL/min. Plasma concentrations fell by more than 95% within 6 hours following IV administration. The terminal half-life in humans is estimated to exceed 10 years, reflecting release of alendronate from the skeleton.
Cholecalciferol: When radioactive vitamin D
3 was administered to healthy subjects, the mean urinary excretion of radioactivity after 48 hours was 2.4%, and the mean fecal excretion of radioactivity after 4 days was 4.9%. In both cases, the excreted radioactivity was almost exclusively as metabolites of the parent. The mean half-life of vitamin D
3 in the serum following an oral dose of FOSAMAX PLUS (70 mg/2800 IU) is approximately 24 hours.
Characteristics in Patients: Preclinical studies show that the alendronate that is not deposited in bone is rapidly excreted in the urine. No evidence of saturation of bone uptake was found after chronic dosing with cumulative IV doses up to 35 mg/kg in animals. Although no clinical information is available, it is likely that, as in animals, elimination of alendronate via the kidney will be reduced in patients with impaired renal function. Therefore, somewhat greater accumulation of alendronate in bone might be expected in patients with impaired renal function (see DOSAGE & ADMINISTRATION).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image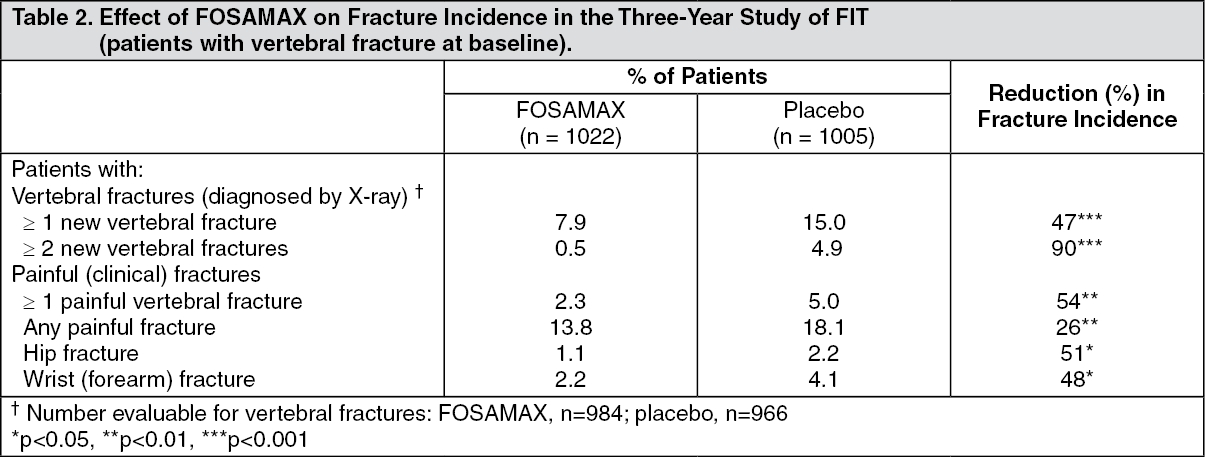 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image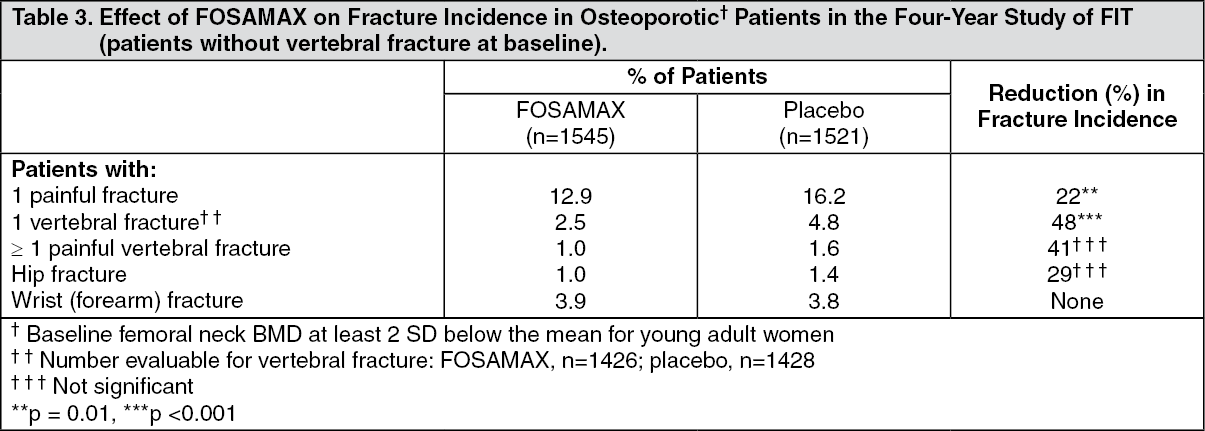 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image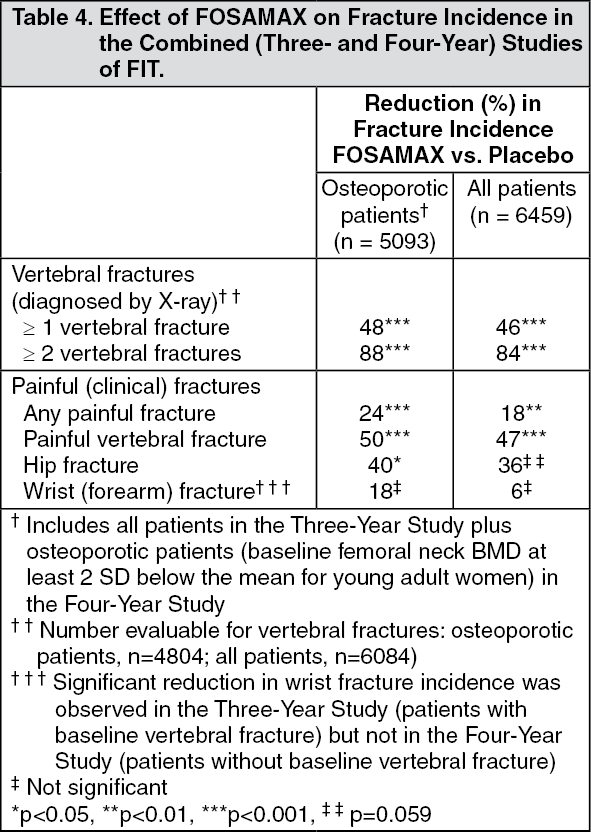 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
