Clinical Trial Data: Adverse Reactions Reported by ≥ 1% of Tramadol hydrochloride + Paracetamol (Dolcet mini)-treated Patients in Placebo controlled Clinical Trials of Tramadol hydrochloride + Paracetamol (Dolcet mini). (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Adverse Reactions Reported by <1% of Ultracet-treated Patients in Placebo controlled Clinical Trials of Tramadol hydrochloride + Paracetamol (Dolcet mini). (See Table 3.)
Click on icon to see table/diagram/image
Adverse Reactions Identified in Clinical Trials and/or Postmarketing Experience with Tramadol. (See Table 4.)
Click on icon to see table/diagram/image
Post-marketing adverse events of Paracetamol Allergic reactions (mainly skin rash) or secondary hypersensitivity reaction to paracetamol were rarely reported, but generally they were recovered after administration was discontinued, and symptomatic treatment was conducted, if necessary. Hematologic disorders including thrombocytopenia and agranulocytosis have been reported, but they are not always related to paracetamol. There were several reports suggesting that a combined use of paracetamol and warfarin similar analogue may result in hypoprothrombinemia. No prothrombin time was altered in other studies.
Post-marketing adverse events: Adverse reactions identified during postmarketing experience: Hyponatremia/syndrome of inappropriate antidiuretic hormone, Fixed eruption.
As a result of post-marketing surveillance in 37,967 patients for 6 years to re-review locally, regardless of the causal relationship with drugs, the case incidence rate was reported as 4.57% (1,737 subjects/37,967 subjects, 2,501 cases). The most common adverse reactions were digestive system disorders such as nausea, constipation and vomiting, etc. with 1,391 cases (3.66%) and it was found that there were 563 cases of adverse event reports regarding nervous system disorders such as dizziness and headache (1.48%), 149 cases of generalized disorders (0.39%), 149 cases of psychiatric disorders (0.39%), 105 cases of skin and appendages disorders (0.28%), 31 cases of blood system disorders (0.08%), 28 cases of kidney and urinary system disorders (0.07%), 28 cases of circulatory system disorders (0.07%), 21 cases of respiratory system disorders (0.06%), 1 case of cardiovascular system disorders and 43 cases of other disorders (0.11%).
The incidence of adverse drug reactions for which causal relationship with this drug could not be ruled out was 4.14% (1,573 subjects/37,967 subjects, 2,244 cases). Major adverse events were nausea 1.49% (567 subjects/37,967 subjects), dizziness 0.95% (358 subjects/37,967 subjects), and others are less than 1% among reported adverse events. The classification of them based on their sites is as follows:
Systemic disorders: Asthenia, Fatigue, Pyrexia, Edema, Chest pain, Spasticity, Syncope.
Cardiovascular system: Congestive heart failure.
Nervous system: Dizziness, Headache, Vertigo, Stupor, Migraine, Tremor, Sensory anomaly, Hypertonia, Exacerbated migraine, Contraction involuntary muscle, and Convulsion.
Respiratory system: Dyspnea, Asthma, Exertional dyspnea, Cough, and Bronchitis.
Kidney and urinary system: Dysuria, Urinary retention, Pollakiuria, Abnormal value of renal function and Oliguria.
Skin and appendages: Pruritus, Rash, Diaphoresis, Urticaria.
Digestive system: Nausea, Vomiting, Dyspepsia, Stomachache, Constipation, Dry mouth, Diarrhea, Dysphagia, Flatus, Tongue edema, Increased saliva, Progression of rectal cancer, Progression of esophageal cancer, Progression of gastric cancer, Paralytic ileus, Hiccup and Cholecystitis.
Psychiatric system: Drowsiness, Insomnia, Anorexia, Anxiety, Depression, Nervousness, Mood instability, Hallucination, and Depersonalization.
Blood system: Anemia, Oligochromemia, Erythrocytopenia, Leukocytopenia, Leukocytosis and Thrombocytopenia.
Circulatory system: Palpitation, Hypertension, Arrhythmia, Tachycardia, Aggravated hypertension, Aortostenosis.
Other: Melena, Increased cholesterol, Weight loss, Tinnitus, Chills, Buttock pain, Femoral pain, Allergic rhinitis, Epistaxis, Abnormal visual field, Progression of biliary tract cancer, Foreign body sensation in the eyes, Progression of liver cancer, and Hepatic dysfunction.
Serious adverse drug reactions were reported 1 case of hepatic dysfunction, cholecystitis, paralytic ileus and stupor, respectively, and unexpected adverse drug reactions were reported 1 case of cholecystitis, hiccup, paralytic ileus, abnormal renal function count (increased BUN/CR), increased saliva and melena, respectively.
The incidence rate of adverse drug reactions occurred in the study in patients with renal disorders, who are special patients, was 6.25% (11 pts/176 pts, 165 cases), a major adverse drug reaction was 4 cases of nausea and vertigo, respectively (2.27%). There were no serious adverse drug reactions and unexpected adverse events. The incidence rate of adverse drug reactions occurred in the study in patients with hepatic disorders, who are special patients, was 6.63 % (22 pts/332 pts, 36 cases), and major adverse drug reactions were 12 cases of nausea (3.61%), 6 cases of vertigo (1.81%). Serious adverse drug reactions were 1 case of paralytic ileus, cholecystitis and hepatic dysfunction, respectively (0.30%), and unexpected adverse reactions were 1 case of paralytic ileus and cholecystitis, respectively.
Serious adverse reactions which were reported voluntarily during local post-marketing surveillance were total 23 cases; 3 cases of nausea, 2 cases of dizziness and dyspnea and 1 case of stupor, migraine, language disorders, unconsciousness, recurrence of myocardial infarction, hypotension, vomiting, dyspepsia, confusion, multiple organ failure, anaphylaxis, and intentional overdose, respectively. This was reported from the uncertain size of population, and it is difficult to estimate the frequency and the causal relation of this drug.
And as a result of post-marketing clinical tests conducted on total of 5,566 people separately from post-marketing surveillance during the post-marketing survey period in Korea, total 3 cases of unexpected adverse drug reactions including 2 cases of eructation (0.04%), 1 case of chest discomfort (0.02%) were reported.
The incident of adverse events in patient group with the history of administering therapeutic drugs (anodyne) before administering this drug was shown statistically higher than those without administration history (5.92% vs 3.75%, p<0.001). In addition, in subjects whose mean dosage per day is 4 tabs or more, the incidence rate of adverse reactions was shown statistically and significantly higher than the patient group who received 2 or more and less than 4 tabs (4.36%, (1,325/30,419 pts), p<0.001).



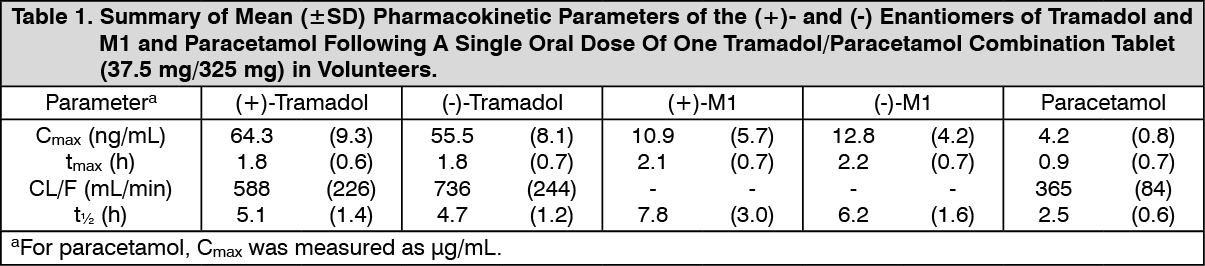 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image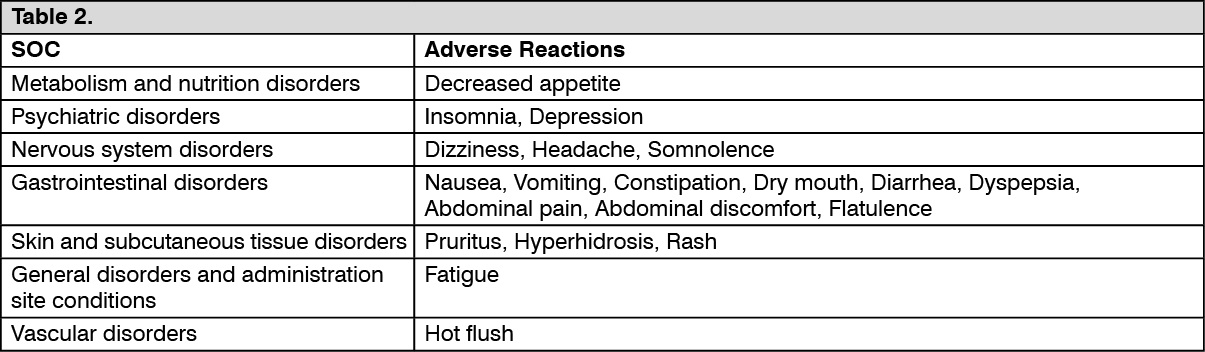 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image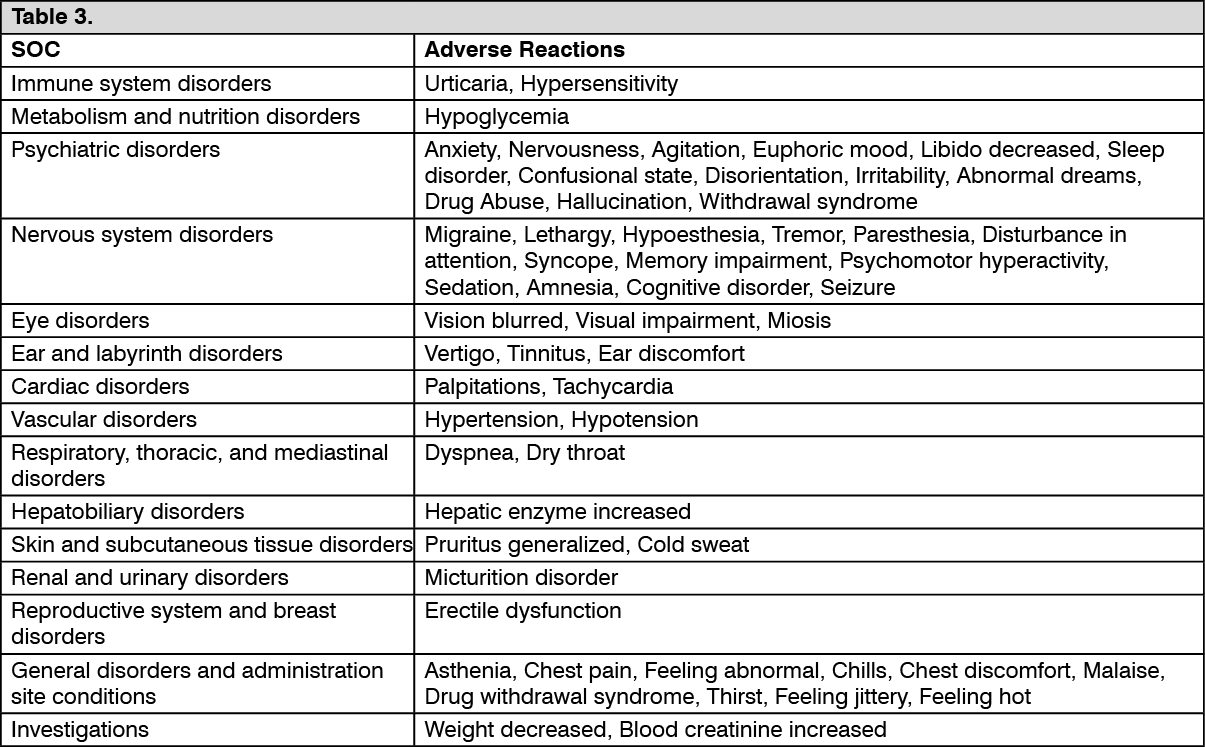 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image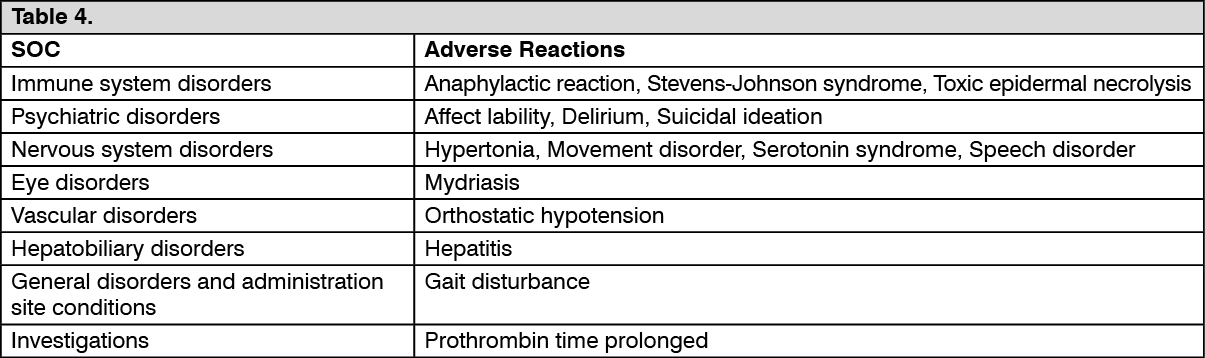 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out