Myelosuppression: The most frequent serious consequence of treatment with Busulfan (IV Busulfex) at the recommended dose and schedule is prolonged myelosuppression, occurring in all patients (100%). Severe granulocytopenia, thrombocytopenia, anemia, or any combination thereof may develop. Hematopoietic progenitor cell transplantation is required to prevent potentially fatal complications of the prolonged myelosuppression. Monitor complete blood counts, including white blood cell differentials, and quantitative platelet counts daily during treatment and until engraftment is demonstrated. Absolute neutrophil counts dropped below 0.5x10
9/L at a median of 4 days post-transplant in 100% of patients treated in the Busulfan (IV Busulfex) clinical trial. The absolute neutrophil count recovered at a median of 13 days following allogeneic transplantation when prophylactic G-CSF was used in the majority of patients. Thrombocytopenia (less than 25,000/mm
3 or requiring platelet transfusion) occurred at a median of 5-6 days in 98% of patients. Anemia (hemoglobin less than 8.0 g/dL) occurred in 69% of patients. Use antibiotic therapy and platelet and red blood cell support when medically indicated.
Seizures: Seizures have been reported in patients receiving high-dose oral busulfan at doses producing plasma drug levels similar to those achieved following the recommended dosage of Busulfan (IV Busulfex). Despite prophylactic therapy with phenytoin, one seizure (1/42 patients) was reported during an autologous transplantation clinical trial of Busulfan (IV Busulfex). This episode occurred during the cyclophosphamide portion of the conditioning regimen, 36 hours after the last Busulfan (IV Busulfex) dose. Initiate phenytoin therapy or any other alternative anti-convulsant prophylactic therapy (e.g., benzodiazepines, valproic acid or levetiracetam) prior to Busulfan (IV Busulfex) treatment (see Dosage & Administration). Use caution when administering the recommended dose of Busulfan (IV Busulfex) to patients with a history of a seizure disorder or head trauma or who are receiving other potentially epileptogenic drugs.
Hepatic Veno-Occlusive Disease (HVOD): Current literature suggests that high busulfan area under the plasma concentration verses time curve (AUC) values (greater than 1,500 µM·min) may be associated with an increased risk of developing HVOD. Patients who have received prior radiation therapy, greater than or equal to three cycles of chemotherapy, or a prior progenitor cell transplant may be at an increased risk of developing HVOD with the recommended Busulfan (IV Busulfex) dose and regimen. Based on clinical examination and laboratory findings, HVOD was diagnosed in 8% (5/61) of patients treated with Busulfan (IV Busulfex) in the setting of allogeneic transplantation, was fatal in 2/5 cases (40%), and yielded an overall mortality from HVOD in the entire study population of 2/61 (3%).Three of the five patients diagnosed with HVOD were retrospectively found to meet the Jones' criteria. The incidence of HVOD reported in the literature from the randomized, controlled trials was 7.7%-12% (see Pharmacology: Pharmacodynamics: Clinical Studies under Actions). Monitor serum transaminases, alkaline phosphatase, and bilirubin daily through BMT Day +28 to detect hepatotoxicity, which may herald the onset of HVOD.
Embryo-fetal Toxicity: Busulfan (IV Busulfex) can cause fetal harm when administered to a pregnant woman based on animal data. Busulfan was teratogenic in mice, rats, and rabbits. The solvent, DMA, may also cause fetal harm when administered to a pregnant woman based on findings in animals. Advise pregnant women of the potential risk to a fetus. Advise females and males of reproductive potential to use effective contraception during and after treatment with Busulfan (IV Busulfex) (see Pregnancy & Lactation).
Cardiac Tamponade: Cardiac tamponade has been reported in pediatric patients with thalassemia (8/400 or 2% in one series) who received high doses of oral busulfan and cyclophosphamide as the preparatory regimen for hematopoietic progenitor cell transplantation. Six of the eight children died and two were saved by rapid pericardiocentesis. Abdominal pain and vomiting preceded the tamponade in most patients. Monitor for signs and symptoms, promptly evaluate and treat if cardiac tamponade is suspected.
Bronchopulmonary Dysplasia: Bronchopulmonary dysplasia with pulmonary fibrosis is a rare but serious complication following chronic busulfan therapy. The average onset of symptoms is 4 years after therapy (range 4 months to 10 years).
Cellular Dysplasia: Busulfan may cause cellular dysplasia in many organs. Cytologic abnormalities characterized by giant, hyperchromatic nuclei have been reported in lymph nodes, pancreas, thyroid, adrenal glands, liver, lungs and bone marrow. This cytologic dysplasia may be severe enough to cause difficulty in the interpretation of exfoliative cytologic examinations of the lungs, bladder, breast and the uterine cervix.
Use in Children: The effectiveness of Busulfan (IV Busulfex) in the treatment of CML has not been specifically studied in pediatric patients. An open-label, uncontrolled study evaluated the pharmacokinetics of Busulfan (IV Busulfex) in 24 pediatric patients receiving Busulfan (IV Busulfex) as part of a conditioning regimen administered prior to hematopoietic progenitor cell transplantation for a variety of malignant hematologic (N=15) or non-malignant diseases (N=9). Patients ranged in age from 5 months to 16 years (median 3 years). Busulfan (IV Busulfex) dosing was targeted to achieve an area under the plasma concentration curve (AUC) of 900-1350 µM·min with an initial dose of 0.8 mg per kg or 1.0 mg per kg (based on ABW) if the patient was greater than 4 or less than or equal to 4 years, respectively. The dose was adjusted based on plasma concentration after completion of dose 1.
Patients received Busulfan (IV Busulfex) doses every six hours as a two-hour infusion over four days for a total of 16 doses, followed by cyclophosphamide 50 mg per kg once daily for four days. After one rest day, hematopoietic progenitor cells were infused. All patients received phenytoin as seizure prophylaxis. The target AUC (900-1350±5% µM·min) for Busulfan (IV Busulfex) was achieved at dose 1 in 71% (17/24) of patients. Steady state pharmacokinetic testing was performed at dose 9 and 13. Busulfan (IV Busulfex) levels were within the target range for 21 of 23 evaluable patients.
All 24 patients experienced neutropenia (absolute neutrophil count less than 0.5x10
9/L) and thrombocytopenia (platelet transfusions or platelet count less than 20,000/mm
3). Seventy-nine percent (19/24) of patients experienced lymphopenia (absolute lymphocyte count less than 0.1x10
9). In 23 patients, the ANC recovered to greater than 0.5x10
9/L (median time to recovery = BMT day +13; range = BMT day +9 to +22). One patient who died on day +20 had not recovered to an ANC >0.5x10
9/L.
Four (17%) patients died during the study. Two patients died within 28 days of transplant; one with pneumonia and capillary leak syndrome, and the other with pneumonia and veno-occlusive disease. Two patients died prior to day 100; one due to progressive disease and one due to multi-organ failure.
Adverse reactions were reported in all 24 patients during the study period (BMT day -10 through BMT day +28) or post-study surveillance period (day +29 through +100). These included vomiting (100%), nausea (83%), stomatitis (79%), HVOD (21%), graft-versus host disease (GVHD) (25%), and pneumonia (21%).
Based on the results of this 24-patient clinical trial, a suggested dosing regimen of Busulfan (IV Busulfex) in pediatric patients is shown in the following dosing nomogram: (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Simulations based on a pediatric population pharmacokinetic model indicate that approximately 60% of pediatric patients will achieve a target Busulfan (IV Busulfex) exposure (AUC) between 900 to 1350 µM·min with the first dose of Busulfan (IV Busulfex) using this dosing nomogram. Therapeutic drug monitoring and dose adjustment following the first dose of Busulfan (IV Busulfex) is recommended.
Dose Adjustment Based on Therapeutic Drug Monitoring: Instructions for measuring the AUC of busulfan at dose 1 (see Blood Sample Collection for AUC Determination as follows) and the formula for adjustment of subsequent doses to achieve the desired target AUC (1125 µM·min), are provided as follows.
Adjusted dose (mg) = Actual Dose (mg) x Target AUC (µM·min)/Actual AUC (µM·min).
For example, if a patient received a dose of 11 mg busulfan and if the corresponding AUC measured was 800 µM·min, for a target AUC of 1125 µM·min, the target mg dose would be: Mg dose =11 mg x 1125 µM·min /800 µM·min =15.5 mg.
Busulfan (IV Busulfex) dose adjustment may be made using this formula and instructions as follows.
Blood Sample Collection for AUC Determination: Calculate the AUC (µM·min) based on blood samples collected at the following time points:
For dose 1:2 hr (end of infusion), 4 hr and 6 hr (immediately prior to the next scheduled Busulfan (IV Busulfex) administration). Actual sampling times should be recorded.
For doses other than dose 1: Pre-infusion (baseline), 2 hr (end of infusion), 4 hr and 6 hr (immediately prior to the next scheduled Busulfan (IV Busulfex) administration).
AUC calculations based on fewer than the three specified samples may result in inaccurate AUC determinations.
For each scheduled blood sample, collect one to three mL of blood into heparinized (Na or Li heparin) Vacutainer tubes. The blood samples should be placed on wet ice immediately after collection and should be centrifuged (at 4°C) within one hour. The plasma, harvested into appropriate cryovial storage tubes, is to be frozen immediately at -20°C. All plasma samples are to be sent in a frozen state (i.e., on dry ice) to the assay laboratory for the determination of plasma busulfan concentrations.
Busulfan (IV Busulfex) AUC calculations may be made using the following instructions and appropriate standard pharmacokinetic formula: Dose 1 AUC
infinity Calculation: AUC
infinity = AUC
0-6hr +AUC
extrapolated, where AUC
0-6hr is to be estimated using the linear trapezoidal rule and AUC extrapolated can be computed by taking the ratio of the busulfan concentration at Hour 6 and the terminal elimination rate constant, λz. The λz must be calculated from the terminal elimination phase of the busulfan concentration vs. time curve. A "0" pre-dose busulfan concentration should be assumed, and used in the calculation of AUC.
If the AUC is assessed subsequent to Dose 1, steady-state AUC
ss (AUC
0-6hr) is to be estimated from the trough, 2 hr, 4 hr and 6 hr concentrations using the linear trapezoidal rule.
Instructions for Drug Administration and Blood Sample Collection for Therapeutic Drug Monitoring: Use an administration set with minimal residual hold up (priming) volume (1-3 mL) for drug infusion to ensure accurate delivery of the entire prescribed dose and to ensure accurate collection of blood samples for therapeutic drug monitoring and dose adjustment.
Prime the administration set tubing with drug solution to allow accurate documentation of the start time of Busulfan (IV Busulfex) infusion. Collect the blood sample from a peripheral IV line to avoid contamination with infusing drug. If the blood sample is taken directly from the existing central venous catheter (CVC), DO NOT COLLECT THE BLOOD SAMPLE WHILE THE DRUG IS INFUSING to ensure that the end of infusion sample is not contaminated with any residual drug. At the end of infusion (2 hr), disconnect the administration tubing and flush the CVC line with 5 cc of normal saline prior to the collection of the end of infusion sample from the CVC port. Collect the blood samples from a different port than that used for the Busulfan (IV Busulfex) infusion. When recording the Busulfan (IV Busulfex) infusion stop time, do not include the time required to flush the indwelling catheter line. Discard the administration tubing at the end of the two-hour infusion (see Caution for Usage).
Use in the Elderly: Clinical studies of Busulfan (IV Busulfex) did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image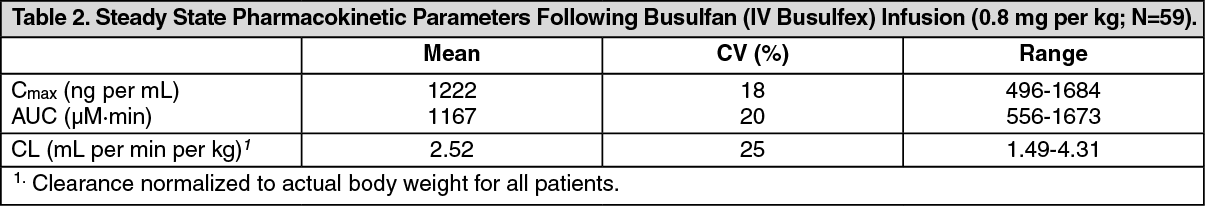 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image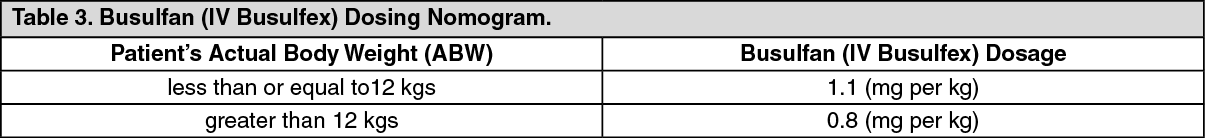 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image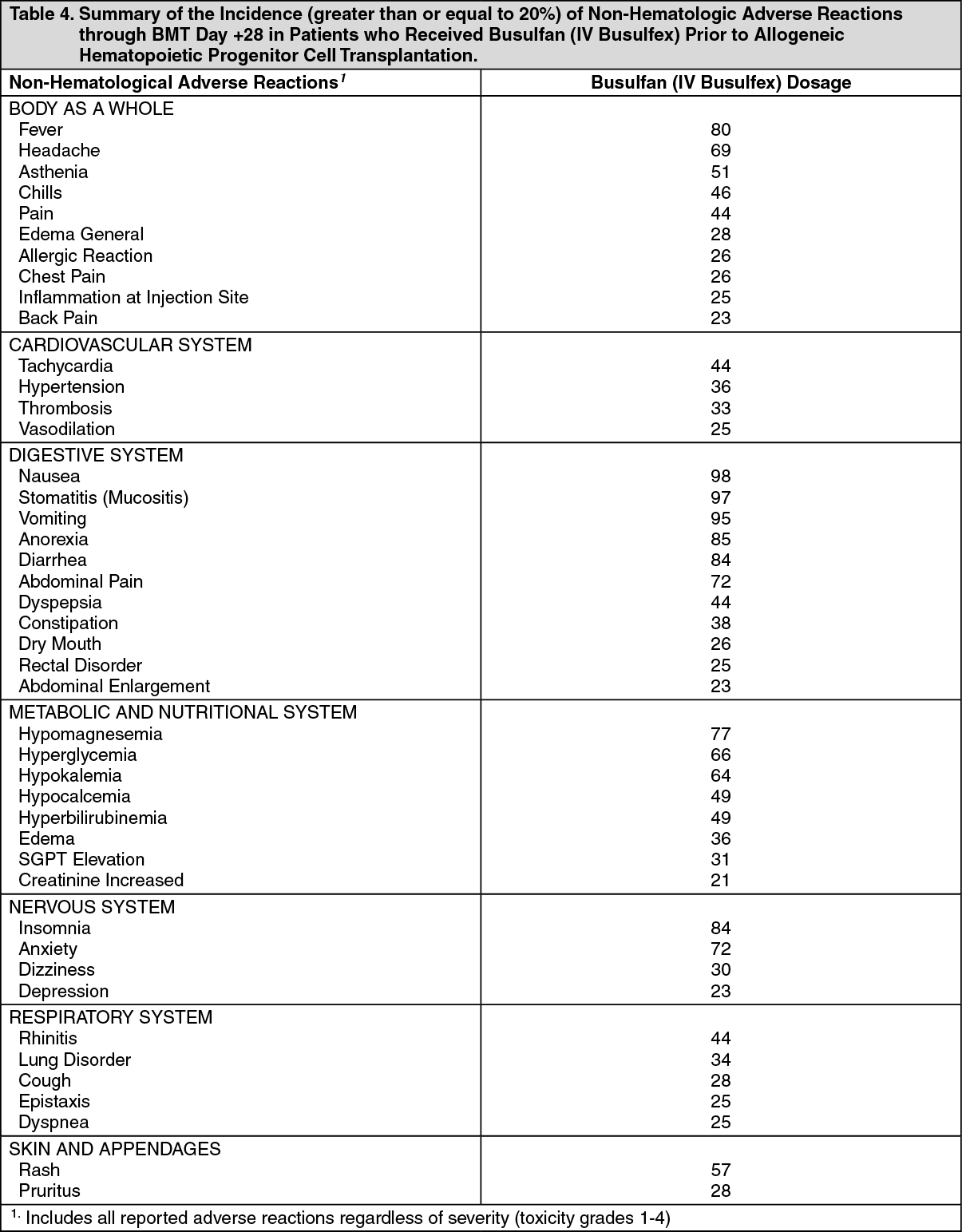 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image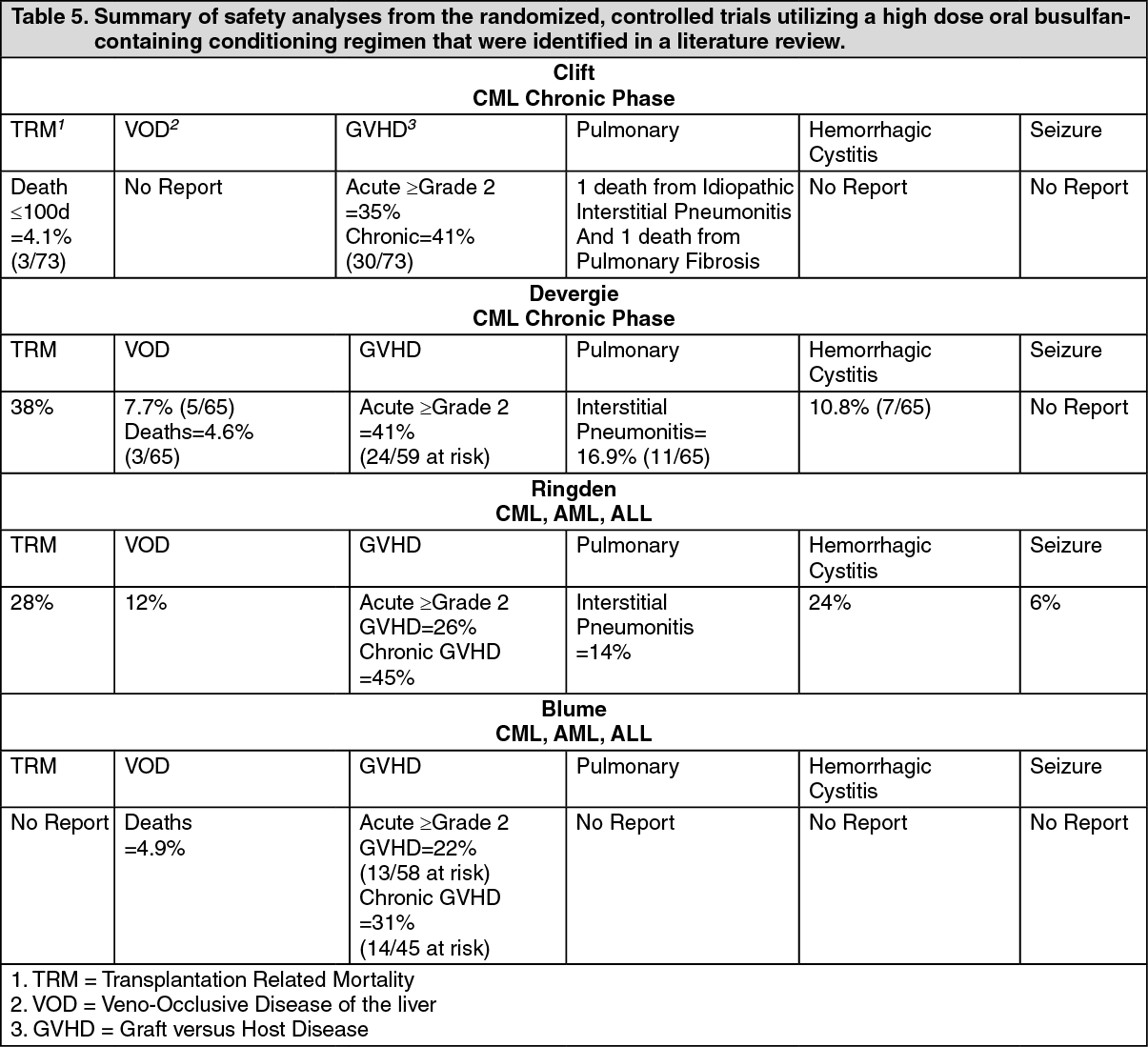 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
