Pharmacology: Pharmacodynamics: Mechanism of action: ADYNOVATE is a full-length recombinant human coagulation factor VIII with an extended half-life. The therapeutic activity of ADYNOVATE is derived from its parent molecule, octocog alfa (ADVATE), which is produced by recombinant DNA technology from a Chinese hamster ovary cell line. The octocog alfa molecule is then covalently conjugated with the PEG reagent, which targets lysine residues. The PEG moiety is conjugated to the octocog alfa molecule to increase the plasma half-life through the reduction of the LRP-1 receptor-mediated clearance of the factor VIII molecule.
Pharmacodynamic effects: The factor VIII/von Willebrand factor complex consists of two molecules (factor VIII and von Willebrand factor) with different physiological functions. When infused into a haemophilic patient, factor VIII binds to von Willebrand factor in the patient's circulation. Activated factor VIII acts as a cofactor for activated factor IX, accelerating the conversion of factor X to activated factor X. Activated factor X converts prothrombin into thrombin. Thrombin then converts fibrinogen into fibrin and a clot can be formed. Haemophilia A is a X-chromosomal linked hereditary disorder of blood coagulation due to decreased levels of factor VIII:C and results in profuse bleeding into joints, muscles or internal organs, either spontaneously or as results of accidental or surgical trauma. By replacement therapy the plasma levels of factor VIII are increased, thereby enabling a temporary correction of the factor deficiency and correction of the bleeding tendencies.
Clinical trials: The safety, efficacy, and PK of ADYNOVATE were evaluated in a multicentre, open-label, prospective, non-randomised, two-arm clinical trial that compared the efficacy of a twice weekly prophylactic treatment regimen to on-demand treatment and determined haemostatic efficacy in the treatment of bleeding episodes. A total of 137 male PTPs (12 to 65 years of age) with severe haemophilia A received at least one infusion with ADYNOVATE. Twenty-five of the 137 subjects were adolescents (12 to less than 18 years of age).
Subjects received either prophylactic treatment (n=120) with ADYNOVATE at a dose of 40-50 IU per kg twice weekly or on-demand treatment (n=17) with ADYNOVATE at a dose of 10-60 IU per kg for a 6-month period. The mean (SD) dose per prophylaxis infusion was 44.4 (3.9) IU per kg with a median dosing interval of 3.6 days. There were 91 out of 98 (93%) subjects previously treated prophylactically prior to enrolment, who experienced a reduction in dosing frequency during routine prophylaxis in the study, with a median reduction of 33.7% (approximately one more day between doses). One hundred eighteen (118) of 120 (98%) prophylaxis subjects remained on the starting recommended regimen without dose adjustment, and 2 subjects increased their dose to 60 IU/kg during prophylaxis due to bleeding in target joints.
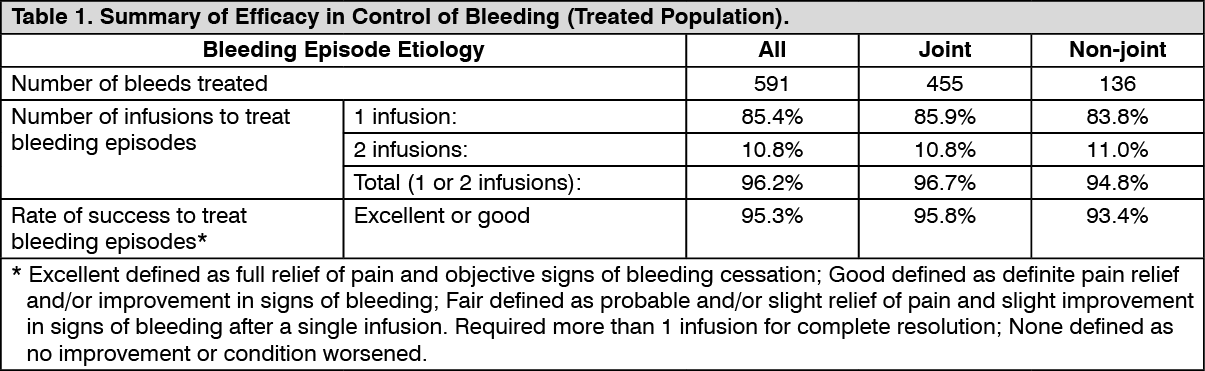
On-Demand Treatment and Control of Bleeding Episodes: A total of 518 bleeding episodes were treated with ADYNOVATE in the per-protocol population, i.e. dosed according to the protocol specific dosing requirements. Of these, 361 bleeding episodes (n=17 subjects) occurred in the on-demand arm and 157 (n=61 subjects) occurred in the prophylaxis arm. The median total dose to treat all bleeding episodes in the per-protocol population was 30.9 (Q1: 21.6; Q3: 45.3) IU per kg. The median dose per infusion to treat all bleeding episodes in the per-protocol population was 29 (Q1: 20.0; Q3: 39.2) IU per kg. The median dose per infusion to treat a minor, moderate, or severe/major bleeding episode in the per-protocol population was 25.5 (Q1: 16.9; Q3: 37.6) IU/kg, 30.9 (Q1: 23.0; Q3: 43.1) IU/kg, or 36.4 (Q1: 29.0; Q3: 44.5) IU/kg, respectively.
A total of 591 bleeding episodes were treated with ADYNOVATE in the treated population, which was identical to the safety analysis set of subjects assigned to routine prophylaxis or on-demand treatment with ADYNOVATE and who received at least one dose of the product. Of these, 361 bleeding episodes (n=17 subjects) occurred in the on-demand arm and 230 bleeding episodes (n=75 subjects) occurred in the routine prophylaxis arm. Efficacy in control of bleeding episodes is summarised in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Routine Prophylaxis: A total of 120 subjects (treated population) received a twice a week regimen in the prophylaxis arm, and an additional 17 subjects were treated episodically in the on-demand arm. In the treated population, the median [mean] annualised bleed rate (ABR) in the on-demand treatment arm was 41.5 [40.8] compared to 1.9 [4.7] while on a twice a week prophylaxis regimen (Table 2). In the per-protocol population, the median [mean] annualised bleed rate (ABR) in the on-demand treatment arm was 41.5 [40.8] compared to 1.9 [3.7] while on a twice a week prophylaxis regimen. Using a negative binomial model to estimate the ABR, there was a significant reduction in the ABR (p<0.0001) for subjects in the prophylaxis arm compared to the on-demand arm. (See Table 2.)
Click on icon to see table/diagram/image
In the treated population, the median [mean] ABR for the 23 adolescent subjects age 12 to <18 years of age on routine prophylaxis was 2.1 [5.2] compared to a median [mean] ABR of 1.9 [4.6] for the 97 subjects 18 years and older. Reduction in ABR between the treatment arms was observed regardless of baseline subgroups examined, including age, presence or absence of target joints, and pre-study treatment regimen. The majority of the bleeding episodes during prophylaxis (95%) were of minor/moderate severity. Forty-five out of 120 subjects (38%) experienced no bleeding episodes and 68 out of 120 subjects (57%) experienced no joint bleeding episodes in the prophylaxis arm. Of those subjects who were compliant to regimen (per-protocol population), 40 out of 101 subjects (40%) experienced no bleeding episodes. All subjects in the on-demand arm experienced a bleeding episode, including a joint bleeding episode.
Routine Prophylaxis Study in Paediatric Subjects (<12 years of age): The safety and efficacy of ADYNOVATE was evaluated in a total of 73 paediatric PTPs with severe haemophilia A, of which 66 subjects were dosed (32 subjects aged <6 years and 34 subjects aged 6 to <12 years) in a separate paediatric study. The prophylactic regimen was 40 to 60 IU/kg of ADYNOVATE twice a week. The median [mean] overall ABR was 2.0 [3.61] for the 66 subjects in the treated population and the median [mean] ABRs for spontaneous and joint bleeding episodes were both 0 [1.18 and 1.12, respectively]. Of the 66 subjects treated prophylactically, 25 (38%) experienced no bleeding episodes, 44 (67%) experienced no spontaneous bleeding episodes, and 48 (73%) experienced no joint bleeding episodes.
Of the 70 bleeding episodes observed during the paediatric study, 82.9% were controlled with 1 infusion and 91.4% were controlled with 1 or 2 infusions. Control of bleeding was rated excellent or good in 63 out of 70 (90%) bleeding episodes. The definitions of excellent or good in the paediatric study were unchanged as compared to the previously conducted prophylaxis study in adolescent and adult subjects.
Long-Term Prophylaxis Treatment in Paediatric and Adult Subjects: The long-term safety and efficacy of ADYNOVATE in prophylaxis and treatment of bleeding episodes was evaluated in 216 paediatric and adult PTPs with severe haemophilia A who had either previously participated in other ADYNOVATE studies or were naïve to ADYNOVATE. In the treated population, subjects received a fixed-dose twice-weekly regimen of 40 to 50 IU/kg if aged ≥12 years or of 40 to 60 IU/kg if aged <12 years. The dose was adjusted up to 80 IU/kg twice weekly if required to maintain factor VIII trough levels of >1%. Subjects that opted for a personalised (pharmacokinetically-tailored) prophylactic regimen received doses up to 80 IU/kg per infusion that targeted factor VIII trough levels of ≥3% at least twice weekly. ABR per prophylactic regimen, bleeding site and etiology are presented in Table 3. (See Table 3.)
Click on icon to see table/diagram/image
Long-term haemostatic efficacy was evaluated in 910 bleeding episodes treated with ADYNOVATE and was rated excellent or good in 88.5% of bleeding episodes. Across age categories and for both the fixed-dose and the PK-tailored dose regimen, >85% of bleed treatments were rated excellent or good. The majority of bleeding episodes were treated with one (74.0%) or two (15.4%) infusions.
Personalised Prophylaxis PROPEL Clinical Trial in Adolescents and Adult Subjects: The safety and efficacy of ADYNOVATE was evaluated in a prospective, randomised, open-label multicentre study in 121 (115 randomised) adolescents (12-18 years old) and adult PTPs with severe haemophilia A for a 12 months treatment period. The study compared 2 PK-guided prophylactic dosing regimens of ADYNOVATE that targeted factor VIII trough levels of 1-3% dosed twice weekly (N=57) or 8-12% dosed every other day (N=58), by assessing the proportions of subjects achieving a total ABR of 0 in the second 6-month study period as the primary efficacy endpoint.
The average prophylactic doses administered in the 1-3% and 8-12% trough arms were 3,866.1 IU/kg per year [mean (SD) infusions/week = 2.3 (0.58)] and 7,532.8 IU/kg per year [mean (SD) infusions/week = 3.6 (1.18)], respectively. After dose adjustment during the first 6-month period of prophylaxis, median trough levels in the second 6-month period (based on the one-stage clotting assay and calculated to the end of the planned infusion interval) ranged from 2.10 IU/dL to 3.00 IU/dL in the 1-3% trough level arm and from 10.70 IU/dL to 11.70 IU/dL in the 8-12% trough level arm, demonstrating that dosing in the 2 prophylaxis regimens was generally adequate to achieve and maintain the desired factor VIII trough levels.
The primary endpoint of the study, proportion of subjects who had a total ABR of 0 during the second 6-month period, was not reached in the ITT patient population (p=0.0545) but was reached in the per-protocol population (nominal p value of 0.0154). The proportions of randomised subjects with total ABRs of 0 during the second 6-month study period and the secondary efficacy outcome measures, spontaneous ABRs and spontaneous annualised joint bleeding rates (AJBRs) of 0 during the second 6-month study period, are presented in Table 4. (See Table 4.)
Click on icon to see table/diagram/image
The secondary efficacy outcome measures (total ABRs, spontaneous ABRs and spontaneous AJBRs during the second 6-month study period), are presented in Table 5. (See Table 5.)
Click on icon to see table/diagram/image
A total of 242 bleeding episodes in 66 subjects were treated with ADYNOVATE; 155 bleeds in 40 subjects in the 1-3% trough level arm and 87 bleeds in 26 subjects in the 8-12% trough level arm. The majority of bleeds (86.0%, 208/242) were treated with 1 or 2 infusions; and bleed treatment at resolution of the bleeding episode was rated excellent or good in 84.7% (205/242) of bleeds.
Perioperative Management Study: A total of 21 major surgical procedures and 5 additional minor surgeries were performed and assessed in 21 unique subjects in the surgery study. For major surgeries, the preoperative loading dose ranged from 36 IU/kg to 109 IU/kg (median: 63 IU/kg); and postoperative total dose ranged from 186 IU/kg to 1320 IU/kg (median: 490 IU/kg). The median total dose for major surgeries was 553 IU/kg (range: 248-1394 IU/kg) and the median total dose of minor surgeries was 106 IU/kg (range: 76-132 IU/kg).
Perioperative haemostatic efficacy was rated as excellent (blood loss less than or equal to that expected for the same type of procedure performed in a non-haemophilic patient, and required blood components for transfusions less than or similar to that expected in non-haemophilic population) for all 26 (21 major, 5 minor) procedures. The median (IQR) observed intraoperative blood loss (n=14) was 10.0 (20.0) mL compared to the predicted average blood loss (n=14) of 150.0 (140.0) mL for major orthopaedic surgeries.
Pharmacokinetics: The pharmacokinetics (PK) of ADYNOVATE was evaluated in a crossover study with ADVATE in 26 subjects (18 adults and 8 adolescents) and in 22 subjects (16 adults and 6 adolescents) after 6 months of treatment with ADYNOVATE. A single dose of 45 ± 5 IU/kg was utilised for both products. In the paediatric study, a single dose of 60 ± 5 IU/kg was utilised for both ADVATE and ADYNOVATE to evaluate PK in 31 paediatrics subjects (<6 years and 6 to <12 years of age). Plasma factor VIII activity was measured by the one-stage clotting assay and chromogenic assay as shown in Table 6 to Table 9. (See Tables 6, 7, 8 and 9.)
ADYNOVATE has an extended half-life of 1.4- to 1.5-fold compared to recombinant full-length human coagulation factor VIII (ADVATE) in the adolescent and adult population, as determined based on one-stage clotting and chromogenic assays, respectively. The half-life extension in the paediatric population was 1.3- to 1.5-fold using both the one-stage clotting and chromogenic assays. An increase in AUC and a decrease in clearance as compared to the parent molecule, ADVATE, were also observed. Incremental recovery was comparable with both products. The change in PK parameters was similar in both the adult and adolescent populations and between one-stage clotting and chromogenic substrate assays.
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Paediatric Pharmacokinetics: Pharmacokinetic parameters calculated from 39 subjects less than 18 years of age (intent-to-treat analysis) are available for 14 children (1 to less than 5 years), 17 older children (6 to less than 12 years) and 8 adolescent subjects (12 to <18 years of age), as shown in Table 8. The mean clearance (based on body weight) of ADYNOVATE was higher and the mean half-life was lower in children less than 12 years of age than adults.
A higher dose may be required in children less than 12 years of age.
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The PK data demonstrates that ADYNOVATE has an extended circulating half-life.
Toxicology: Preclinical safety data: Nonclinical data are limited to 1 month exposure and no studies in juvenile animals were conducted with ADYNOVATE. Thus it was not possible to conclude on the potential risks of PEG accumulation in various tissues/organs relevant for chronic use of ADYNOVATE in the paediatric population.
Genotoxicity: No studies on genotoxicity have been performed with ADYNOVATE.
Carcinogenicity: No studies on carcinogenicity have been performed with ADYNOVATE.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image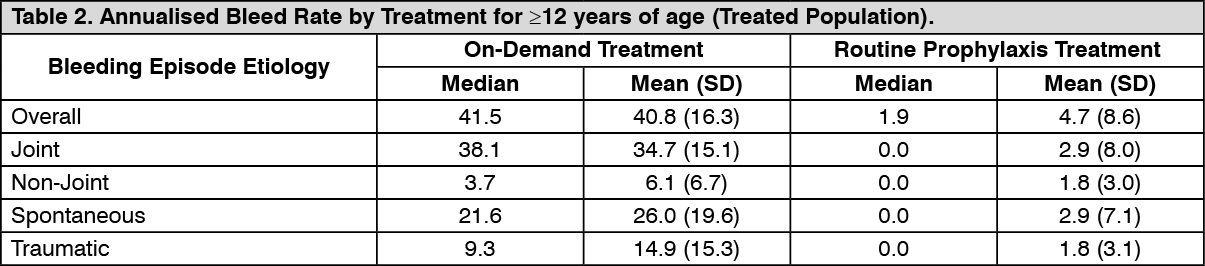 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image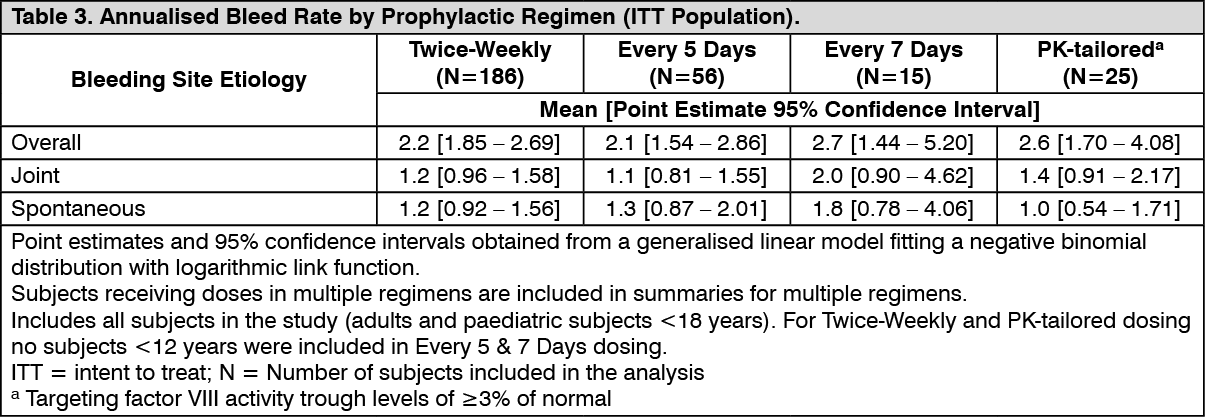 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image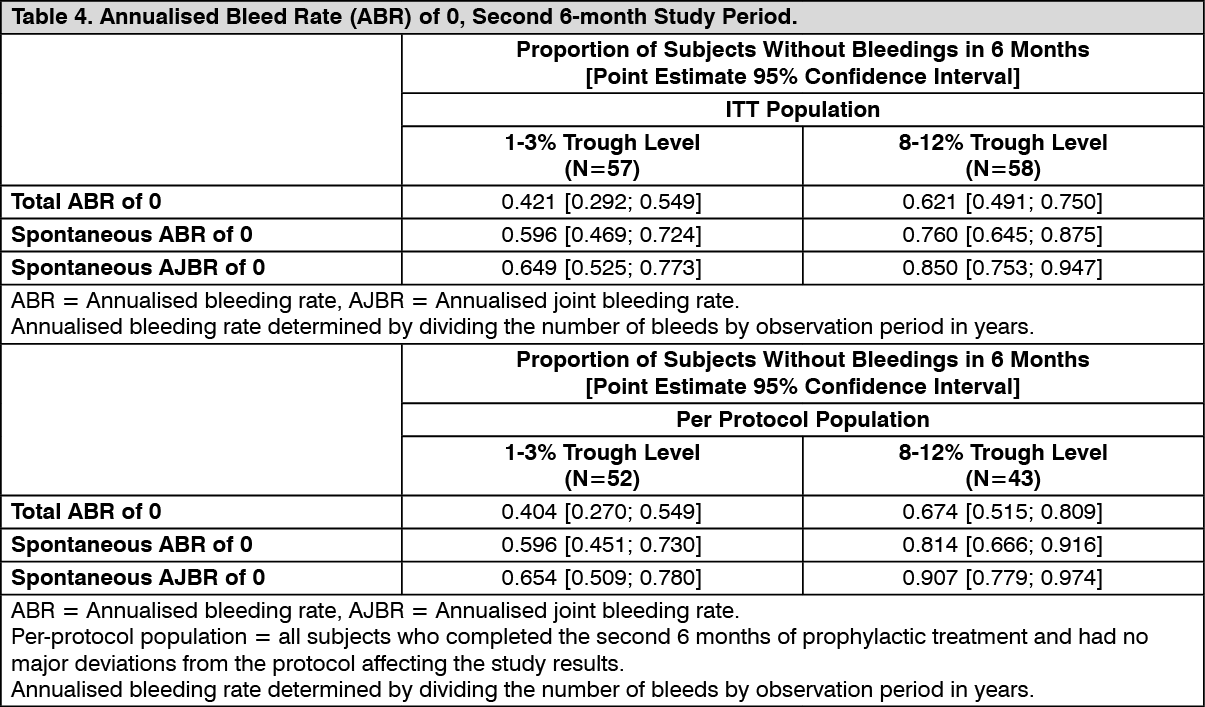 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image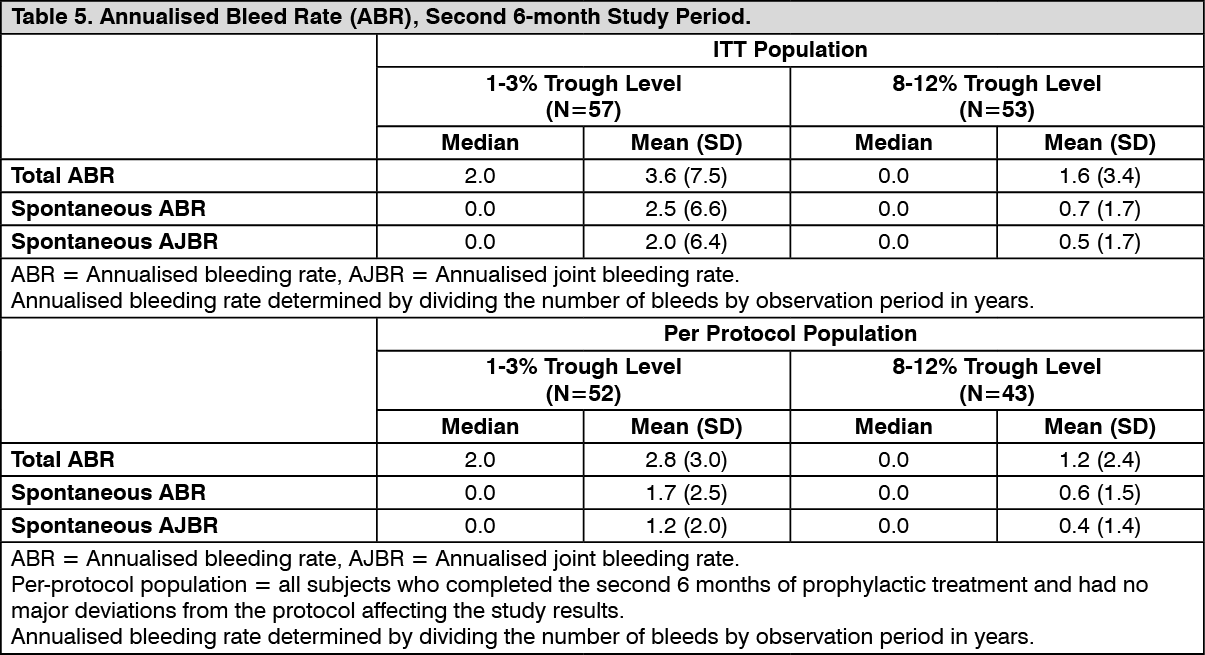 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image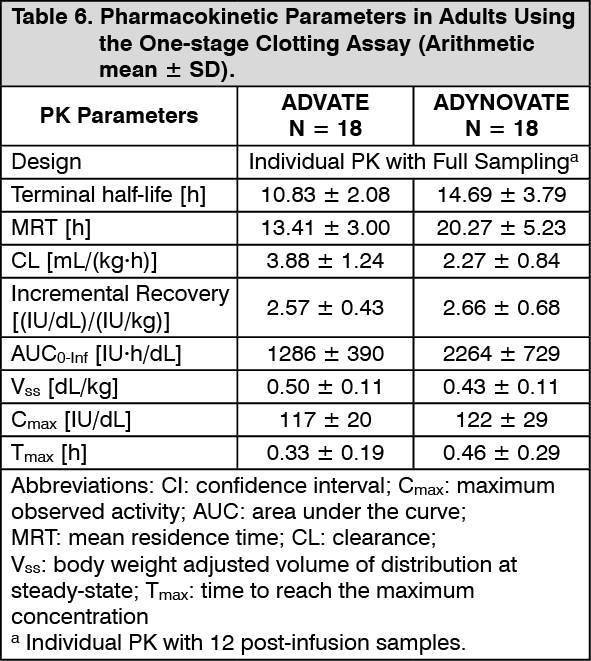 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image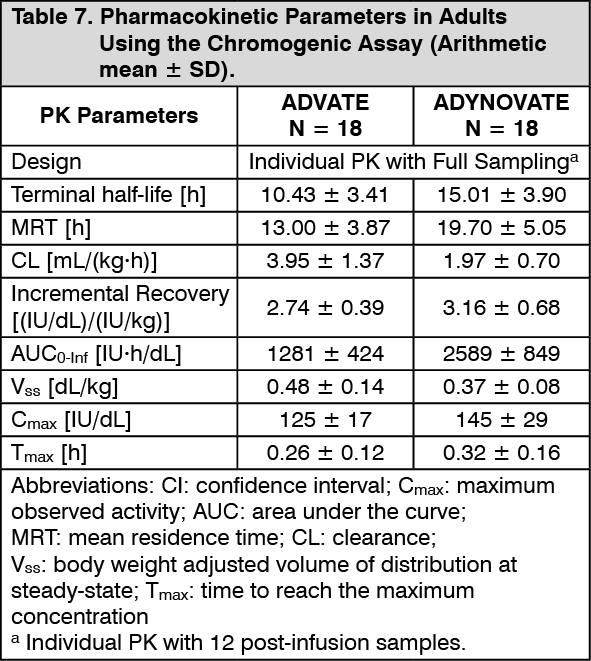 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image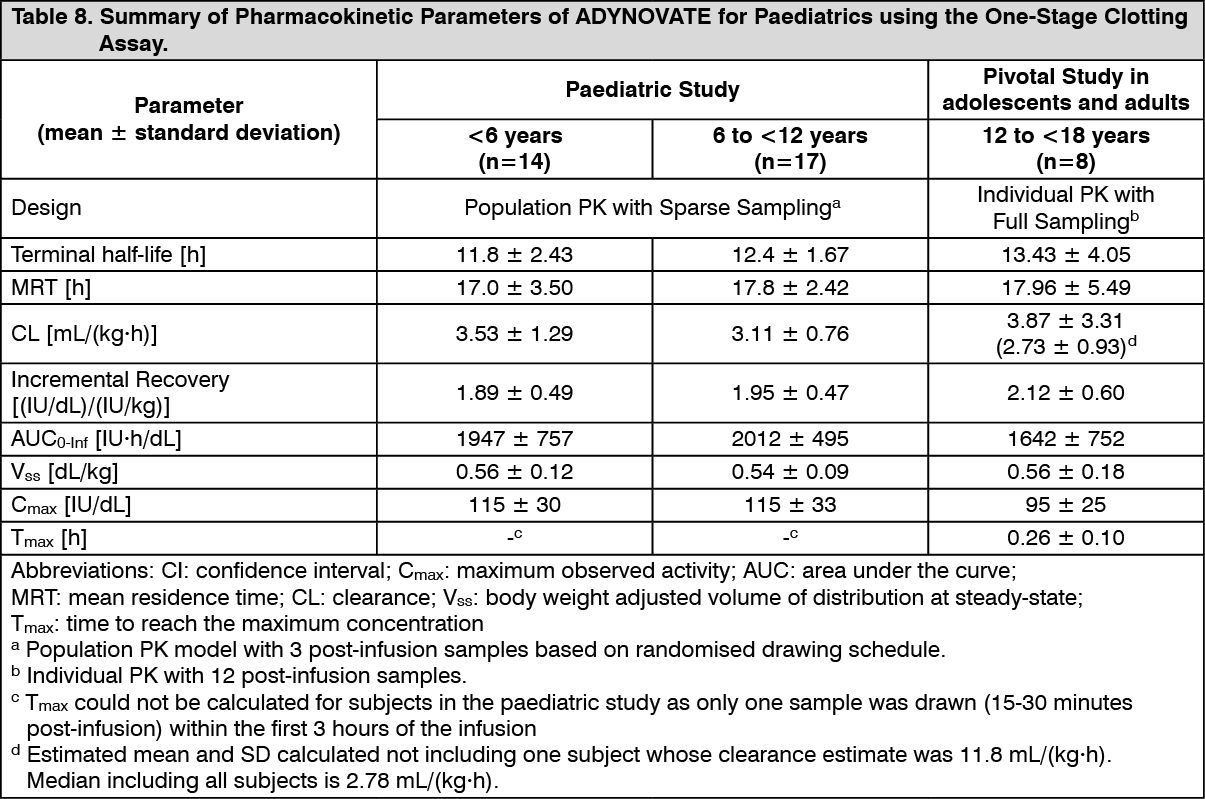 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image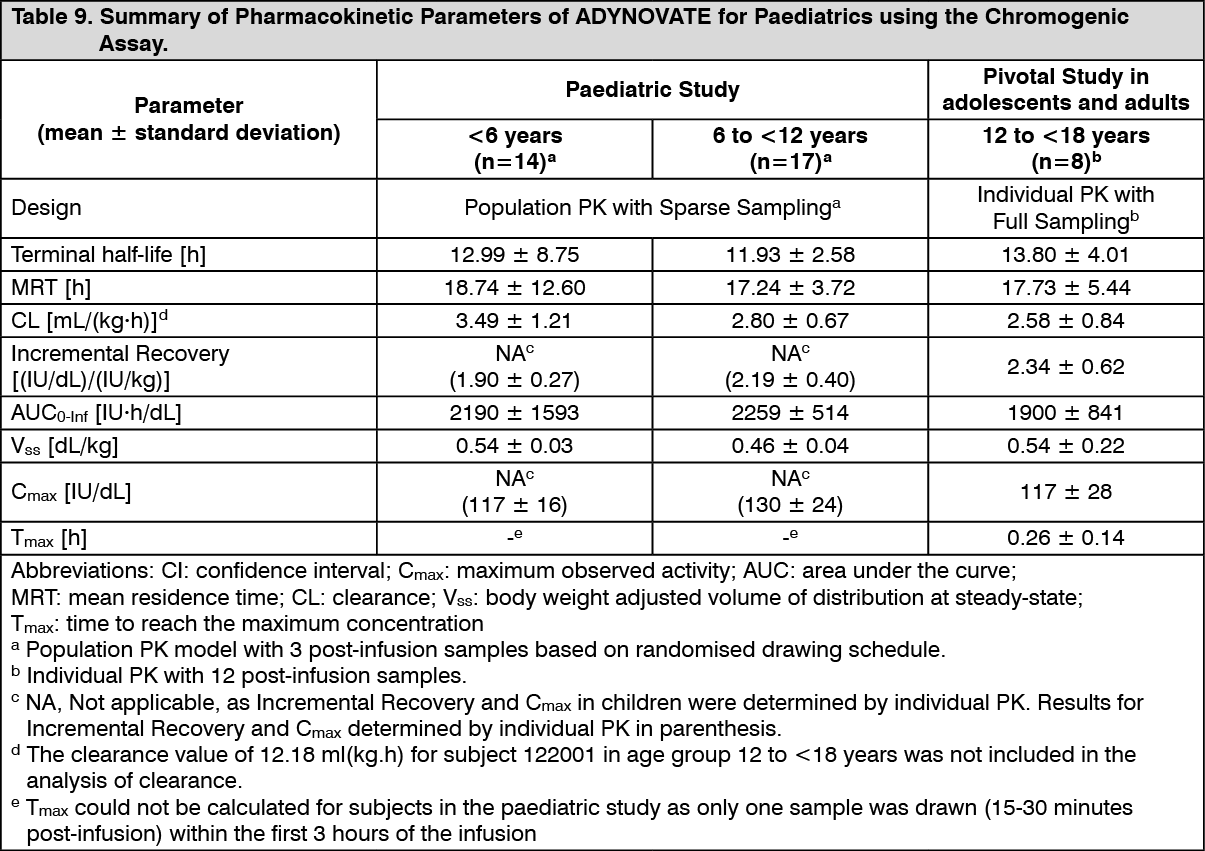 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image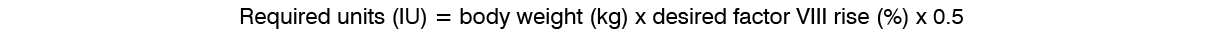 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image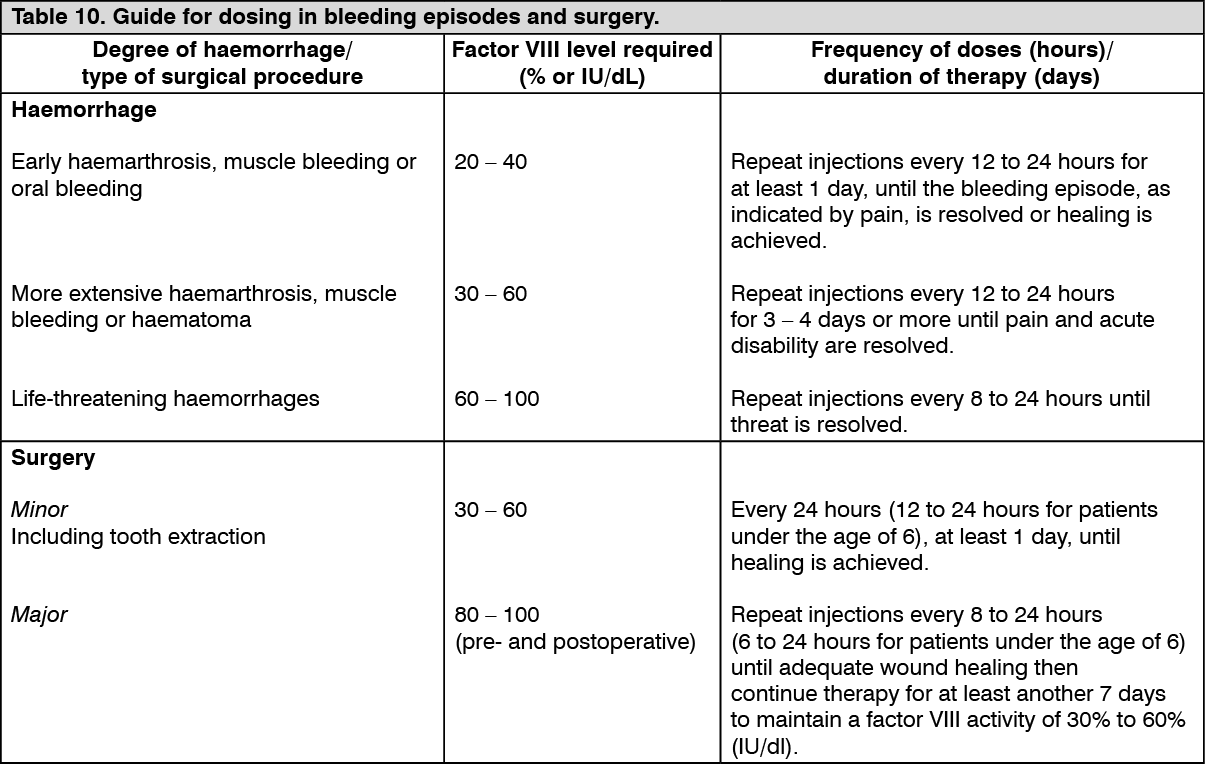 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
