Pharmacology: Mechanism of Action: Erdafitinib is a kinase inhibitor that binds to and inhibits enzymatic activity of FGFR1, FGFR2, FGFR3 and FGFR4 based on
in vitro data. Erdafitinib also binds to RET, CSF1R, PDGFRA, PDGFRB, FLT4, KIT, and VEGFR2. Erdafitinib inhibited FGFR phosphorylation and signaling and decreased cell viability in cell lines expressing FGFR genetic alterations, including point mutations, amplifications, and fusions. Erdafitinib demonstrated antitumor activity in FGFR-expressing cell lines and xenograft models derived from tumor types, including bladder cancer.
Pharmacodynamics: Cardiac Electrophysiology: Based on evaluation of QTc interval in an open-label, dose escalation and dose expansion study in 187 patients with cancer, erdafitinib had no large effect (i.e., > 20 ms) on the QTc interval.
Serum Phosphate: Erdafitinib increased serum phosphate level as a consequence of FGFR inhibition. BALVERSA should be increased to the maximum recommended dose to achieve target serum phosphate levels of 5.5-7.0 mg/dL in early cycles with continuous daily dosing [see Dose Modifications for Adverse Reactions under Dosage & Administration].
In erdafitinib clinical trials, the use of drugs which can increase serum phosphate levels, such as potassium phosphate supplements, vitamin D supplements, antacids, phosphate-containing enemas or laxatives, and medications known to have phosphate as an excipient were prohibited unless no alternatives exist. To manage phosphate elevation, phosphate binders were permitted. Avoid concomitant use with agents that can alter serum phosphate levels before the initial dose increase period based on serum phosphate levels [see Effect of Other Drugs on BALVERSA under Interactions].
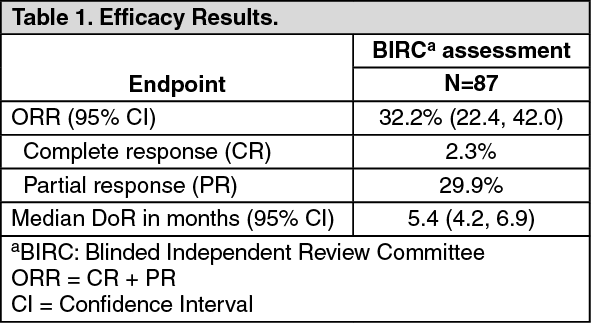
Clinical Studies: Urothelial Carcinoma with Susceptible FGFR Genetic Alterations: Study BLC2001 (NCT02365597) was a multicenter, open-label, single-arm study to evaluate the efficacy and safety of BALVERSA in patients with locally advanced or metastatic urothelial carcinoma (mUC). Fibroblast growth factor receptor (FGFR) mutation status for screening and enrollment of patients was determined by a clinical trial assay (CTA). The efficacy population consists of a cohort of eighty-seven patients who were enrolled in this study with disease that had progressed on or after at least one prior chemotherapy and that had at least 1 of the following genetic alterations: FGFR3 gene mutations (R248C, S249C, G370C, Y373C) or FGFR gene fusions (FGFR3-TACC3, FGFR3-BAIAP2L1, FGFR2-BICC1, FGFR2-CASP7), as determined by the CTA performed at a central laboratory. Tumor samples from 69 patients were tested retrospectively by the QIAGEN therascreen FGFR RGQ RT-PCR Kit, which is the validated test for selection of patients with mUC for BALVERSA.
Patients received a starting dose of BALVERSA at 8 mg once daily with a dose increase to 9 mg once daily in patients whose serum phosphate levels were below the target of 5.5 mg/dL between days 14 and 17; a dose increase occurred in 41% of patients. BALVERSA was administered until disease progression or unacceptable toxicity. The major efficacy outcome measures were objective response rate (ORR) and duration of response (DoR), as determined by blinded independent review committee (BIRC) according to RECIST v1.1.
The median age was 67 years (range: 36 to 87 years), 79% were male, and 74% were Caucasian. Most patients (92%) had a baseline Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Sixty-six percent of patients had visceral metastases. Eighty-four (97%) patients received at least one of cisplatin or carboplatin previously. Fifty-six percent of patients only received prior cisplatin-based regimens, 29% received only prior carboplatin-based regimens, and 10% received both cisplatin and carboplatin-based regimens. Three (3%) patients had disease progression following prior platinum-containing neoadjuvant or adjuvant therapy only. Twenty-four percent of patients had been treated with prior anti PD-L1/PD-1 therapy.
Efficacy results are summarized in Table 1 and Table 2. Overall response rate was 32.2%. Responders included patients who had previously not responded to anti PD-L1/PD-1 therapy. (See Tables 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pharmacokinetics:
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pharmacokinetics: Following administration of 8 mg once daily, the mean (coefficient of variation [CV%]) erdafitinib steady-state maximum observed plasma concentration (C
max), area under the curve (AUC
tau), and minimum observed plasma concentration (C
min) were 1,399 ng/mL (51%), 29,268 ng·h/mL (60%), and 936 ng/mL (65%), respectively.
Following single and repeat once daily dosing, erdafitinib exposure (maximum observed plasma concentration [C
max] and area under the plasma concentration time curve [AUC]) increased proportionally across the dose range of 0.5 to 12 mg (0.06 to 1.3 times the maximum approved recommended dose). Steady state was achieved after 2 weeks with once daily dosing and the mean accumulation ratio was 4-fold.
Absorption: Median time to achieve peak plasma concentration (t
max) was 2.5 hours (range: 2 to 6 hours).
Effect of Food: No clinically meaningful differences with erdafitinib pharmacokinetics were observed following administration of a high-fat and high-calorie meal (800 calories to 1,000 calories with approximately 50% of total caloric content of the meal from fat) in healthy subjects.
Distribution: The mean apparent volume of distribution of erdafitinib was 29 L in patients.
Erdafitinib protein binding was 99.8% in patients, primarily to alpha-1-acid glycoprotein.
Elimination: The mean total apparent clearance (CL/F) of erdafitinib was 0.362 L/h in patients.
The mean effective half-life of erdafitinib was 59 hours in patients.
Metabolism: Erdafitinib is primarily metabolized by CYP2C9 and CYP3A4. The contribution of CYP2C9 and CYP3A4 in the total clearance of erdafitinib is estimated to be 39% and 20% respectively. Unchanged erdafitinib was the major drug-related moiety in plasma, there were no circulating metabolites.
Excretion: Following a single oral dose of radiolabeled erdafitinib, approximately 69% of the dose was recovered in feces (19% as unchanged) and 19% in urine (13% as unchanged).
Specific Populations: No clinically meaningful trends in the pharmacokinetics of erdafitinib were observed based on age (21-88 years), sex, race, body weight (36-132 kg), mild (eGFR [estimated glomerular filtration rate, using modification of diet in renal disease equation] 60 to 89 mL/min/1.73 m
2) or moderate (eGFR 30-59 mL/min/1.73 m
2) renal impairment or mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN, or total bilirubin > 1.0-1.5 x ULN and any AST).
The pharmacokinetics of erdafitinib in patients with severe renal impairment, renal impairment requiring dialysis, moderate or severe hepatic impairment is unknown.
Drug Interaction Studies: Clinical Studies and Model-Based Approaches: Strong CYP2C9 Inhibitors: Erdafitinib mean ratios (90% CI) for C
max and AUC
inf were 121% (99.9, 147) and 148% (120, 182), respectively, when co-administered with fluconazole, a strong CYP2C9 inhibitor and moderate CYP3A4 inhibitor, relative to erdafitinib alone.
Strong CYP3A4 Inhibitors: Erdafitinib mean ratios (90% CI) for C
max and AUC
inf were 105% (86.7, 127) and 134% (109, 164), respectively, when co-administered with itraconazole (a strong CYP3A4 inhibitor and P-gp inhibitor) relative to erdafitinib alone.
Strong CYP3A4/2C9 Inducers: Simulations suggested that rifampicin (a strong CYP3A4/2C9 inducer) may significantly decrease erdafitinib C
max and AUC.
In Vitro Studies: CYP Substrates: Erdafitinib is a time dependent inhibitor and inducer of CYP3A4. The effect of erdafitinib on a sensitive CYP3A4 substrate is unknown. Erdafitinib is not an inhibitor of other major CYP isozymes at clinically relevant concentrations.
Transporters: Erdafitinib is a substrate and inhibitor of P-gp. P-gp inhibitors are not expected to affect erdafitinib exposure to a clinically relevant extent. Erdafitinib is an inhibitor of OCT2.
Erdafitinib does not inhibit BCRP, OATP1B, OATP1B3, OAT1, OAT3, OCT1, MATE-1, or MATE-2K at clinically relevant concentrations.
Acid-Lowering Agents: Erdafitinib has adequate solubility across the pH range of 1 to 7.4. Acid-lowering agents (e.g., antacids, H
2-antagonists, proton pump inhibitors) are not expected to affect the bioavailability of erdafitinib.
Pharmacogenomics: CYP2C9 activity is reduced in individuals with genetic variants, such as the CYP2C9*2 and CYP2C9*3 polymorphisms. Erdafitinib exposure was similar in subjects with CYP2C9*1/*2 and *1/*3 genotypes relative to subjects with CYP2C9*1/*1 genotype (wild type). No data are available in subjects characterized by other genotypes (e.g., *2/*2, *2/*3, *3/*3). Simulation suggested no clinically meaningful differences in erdafitinib exposure in subjects with CYP2C9*2/*2 and *2/*3 genotypes. The exposure of erdafitinib is predicted to be 50% higher in subjects with the CYP2C9*3/*3 genotype, estimated to be present in 0.4% to 3% of the population among various ethnic groups.
Toxicology: Nonclinical Toxicology: Carcinogenesis, Mutagenesis, and Impairment of Fertility: Carcinogenicity studies have not been conducted with erdafitinib.
Erdafitinib was not mutagenic in a bacterial reverse mutation (Ames) assay and was not clastogenic in an
in vitro micronucleus or an
in vivo rat bone marrow micronucleus assay.
Fertility studies in animals have not been conducted with erdafitinib. In the 3-month repeat-dose toxicity study, erdafitinib showed effects on female reproductive organs (necrosis of the ovarian corpora lutea) in rats at an exposure less than the human exposure (AUC) at maximum recommended human dose.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image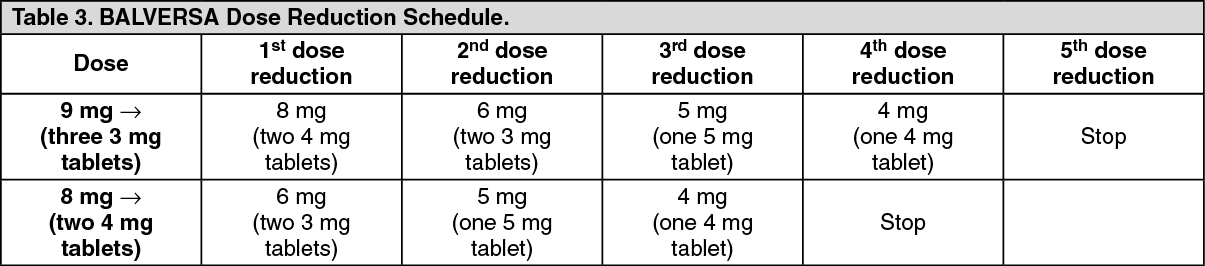 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image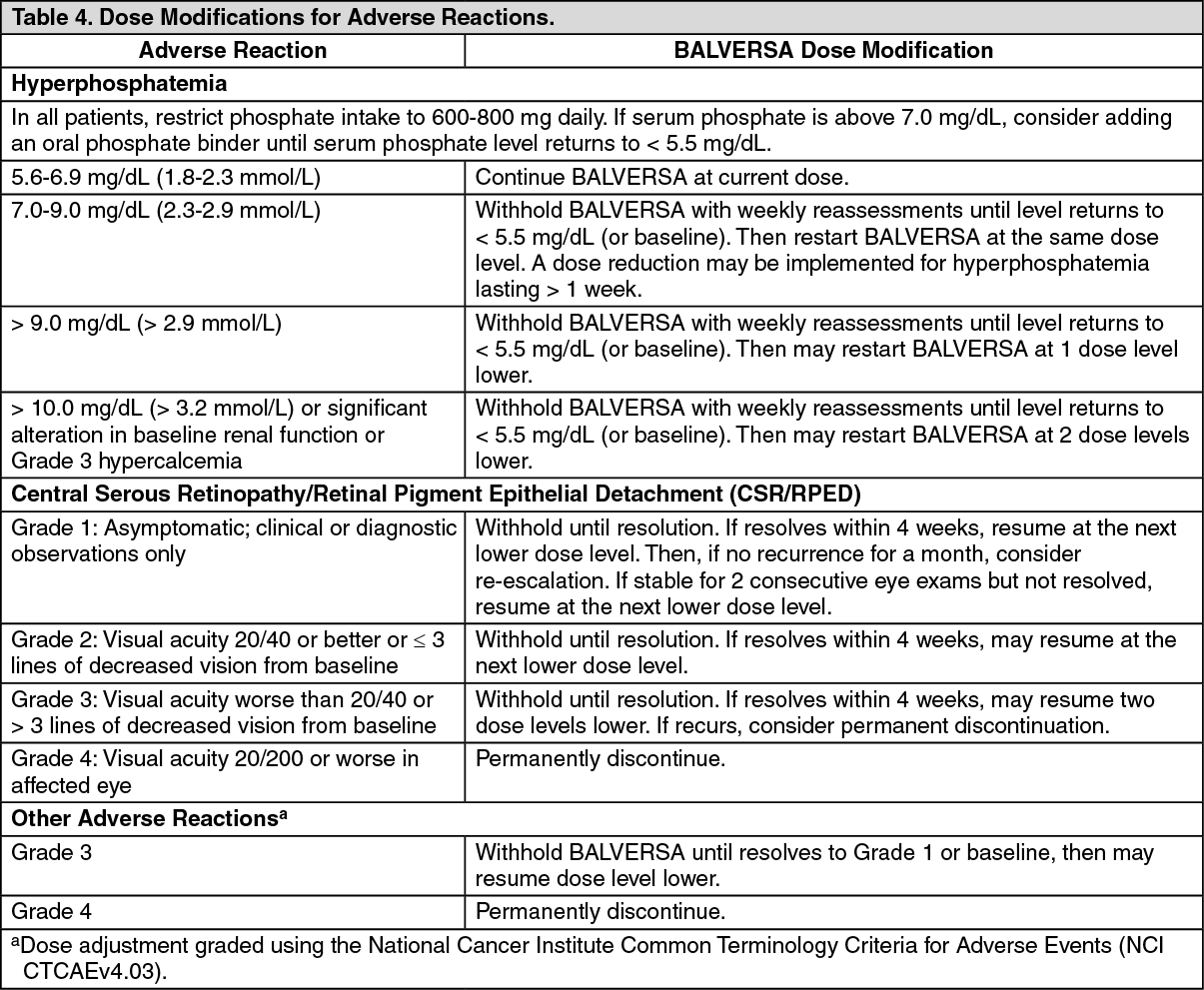 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image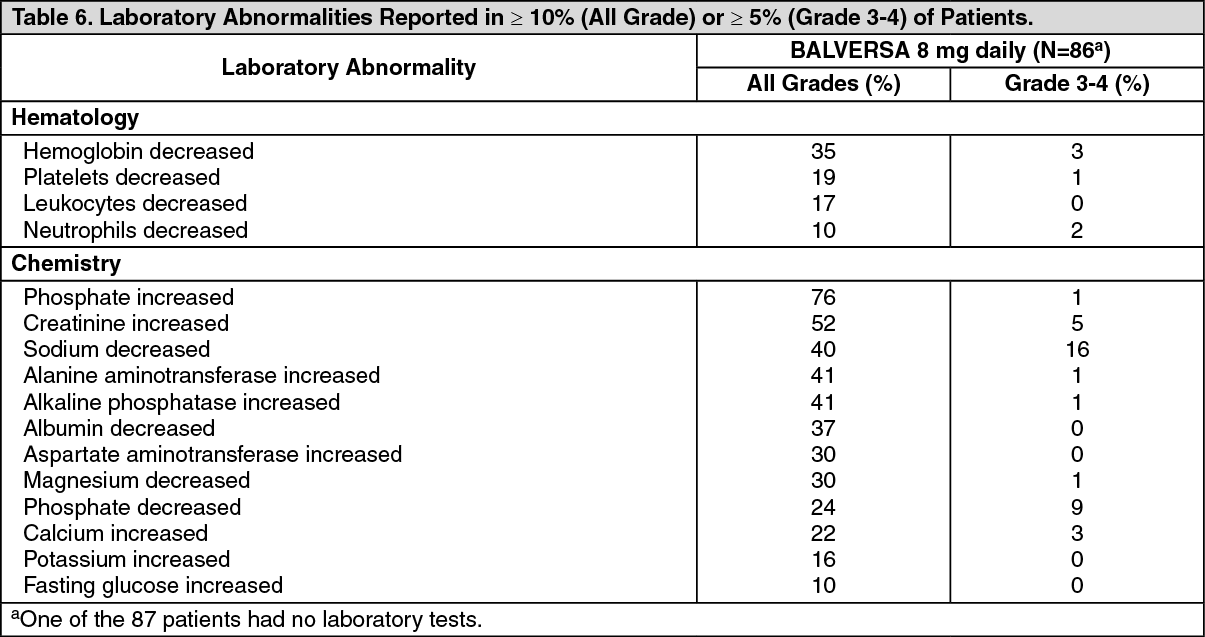 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image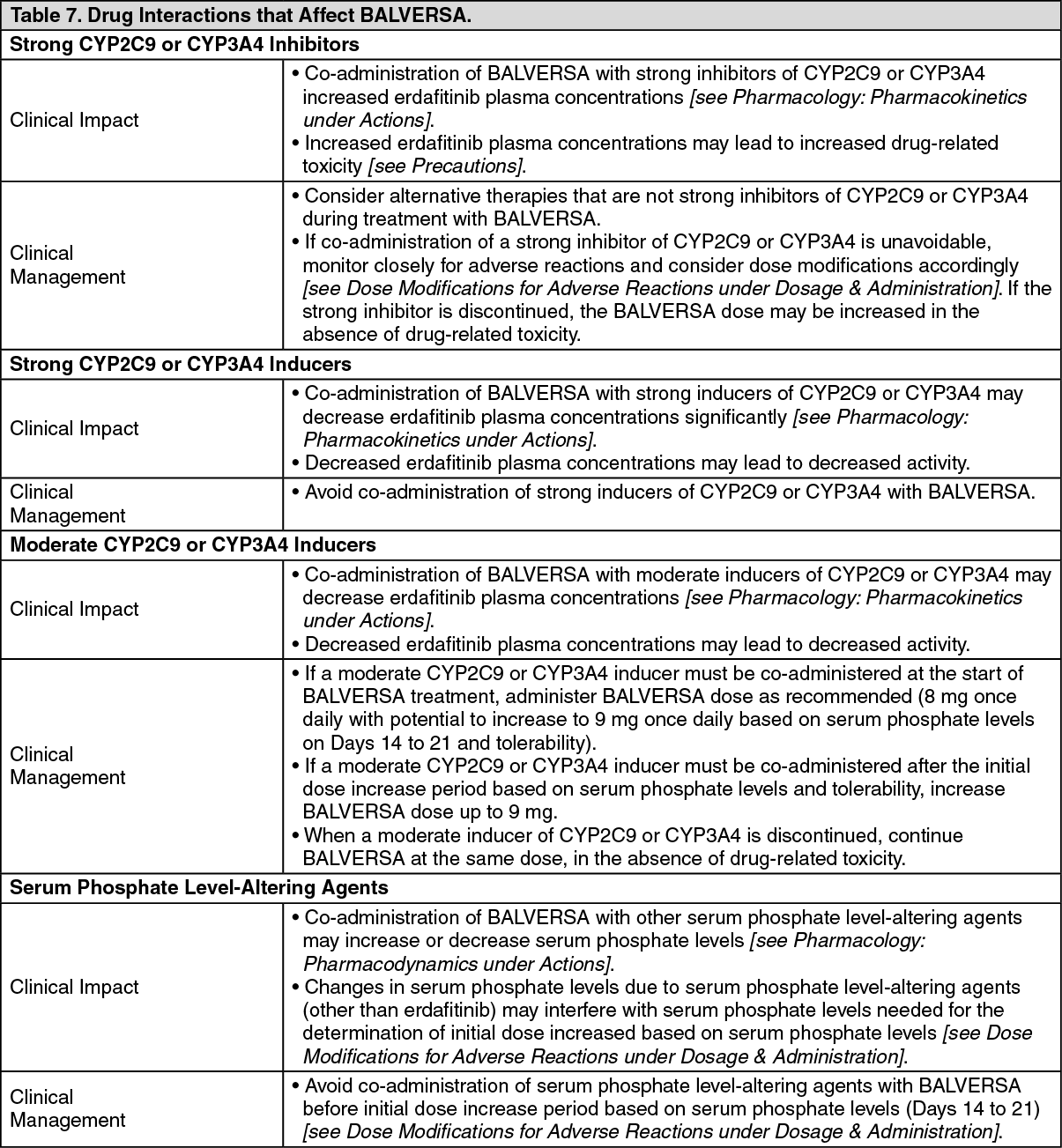 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image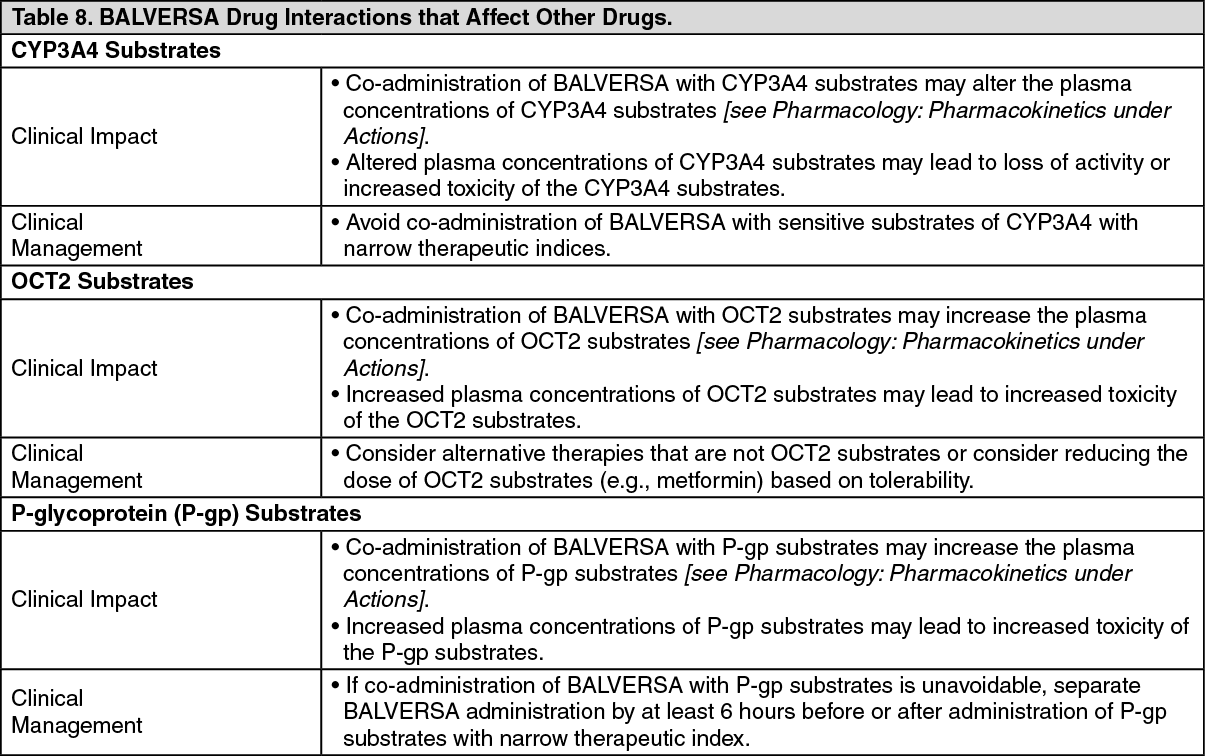 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image



 Sign Out
Sign Out