


 Sign Out
Sign Out
Pharmacology: Pharmacodynamics: Mechanism of action and pharmacodynamic effects: The exact neurochemical appetite suppressant effects of naltrexone/bupropion are not fully understood. The medicinal product has two components: naltrexone, a mu-opioid antagonist, and bupropion, a weak inhibitor of neuronal dopamine and norepinephrine reuptake. These components affect two principal areas of the brain, specifically the arcuate nucleus of the hypothalamus and the mesolimbic dopaminergic reward system.
In the arcuate nucleus of the hypothalamus, bupropion stimulates pro-opiomelanocortin (POMC) neurons that release alpha-melanocyte stimulating hormone (α-MSH), which in turn binds to and stimulates melanocortin 4 receptors (MC4-R). When α-MSH is released, POMC neurons simultaneously release β-endorphin, an endogenous agonist of the mu-opioid receptors. Binding of β-endorphin to mu-opioid receptors on POMC neurons mediates a negative feedback loop on POMC neurons leading to a decrease in the release of α-MSH. Blocking this inhibitory feedback loop with naltrexone is proposed to facilitate a more potent and longer-lasting activation of POMC neurons, thereby amplifying the effects of bupropion on energy balance. Preclinical data suggests that naltrexone and bupropion may have greater than additive effects in this region to reduce food intake when administered together.
Clinical efficacy and safety: The effects of naltrexone/bupropion on weight loss, weight maintenance, waist circumference, body composition, obesity-related markers for cardiovascular and metabolic parameters and patient reported assessments were examined in double-blind, placebo-controlled obesity Phase 2 and Phase 3 trials (BMI range 27-45 kg/m2) with study durations of 16 to 56 weeks randomised to naltrexone hydrochloride (16 to 50 mg/day) and/or bupropion hydrochloride (300 to 400 mg/day) or placebo.
Effect on weight loss and weight maintenance: Four multicentre, double-blind, placebo-controlled obesity Phase 3 studies (NB-301, NB-302, NB-303 and NB-304) were conducted to evaluate the effect of naltrexone/bupropion in conjunction with lifestyle modification in 4,536 subjects randomised to naltrexone/bupropion or placebo. Treatment was initiated with a dose escalation period. Three of these studies (NB-301, NB-302 and NB-304) designated the primary endpoint at 56 weeks, and 1 study (NB-303) designated the primary endpoint at week 28, but continued for 56 weeks. Studies NB-301, NB-303, and NB-304 included periodic instruction from the study sites to reduce caloric intake and increase physical activity, while NB-302 included an intensive behavioral modification program consisting of 28 group counseling sessions over 56 weeks, as well as a prescribed rigorous diet and exercise regimen. NB-304 evaluated subjects with type 2 diabetes not achieving glycaemic goal of HbA1c <7% (53 mmol/mol) with oral anti-diabetes agents or on diet and exercise alone. NB-303 included a re-randomisation in a blinded manner and the addition of a higher dose of naltrexone (naltrexone hydrochloride 48 mg/bupropion hydrochloride 360 mg) at week 28 to half of the cohort of subjects in the active treatment arm who did not adequately respond to treatment, and as such the primary endpoint comparing weight change with 32 mg naltrexone hydrochloride/360 mg bupropion hydrochloride vs. placebo was evaluated at week 28.
Of the overall population of 4,536 subjects in the naltrexone/bupropion Phase 3 studies, 25% had hypertension, 33% had fasting glucose levels ≥100 mg/dL (5.6 mmol/L) at baseline, 54% had dyslipidaemia at study entry, and 11% had type 2 diabetes.
In the combined Phase 3 studies, the mean age was 46 years, 83% were female, and 77% were White, 18% were Black and 5% were other races. Baseline mean BMI was 36 kg/m2 and mean waist circumference was 110 cm. The two co-primary endpoints were percent change from baseline body weight and the proportion of subjects achieving ≥5% total decreased body weight. Data summaries for mean change in body weight reflect the Intent-to-Treat (ITT) population, defined as subjects who were randomised, had a baseline body weight measurement, and had at least one post-baseline body weight measurement during the defined treatment phase, using a last observation carried forward (LOCF) analysis, as well as a completers analysis. Summaries of the proportion of subjects achieving ≥5% or ≥10% reduction in body weight utilise a baseline observation carried forward (BOCF) analysis of all randomised subjects. Overall adherence was similar between trials, and similar between treatment groups. Treatment adherence rates for the integrated Phase 3 studies were: 67% NB vs. 74% placebo at 16 weeks, 63% NB vs. 65% Placebo at 26 weeks, 55% NB vs. 55% placebo at 52 weeks.
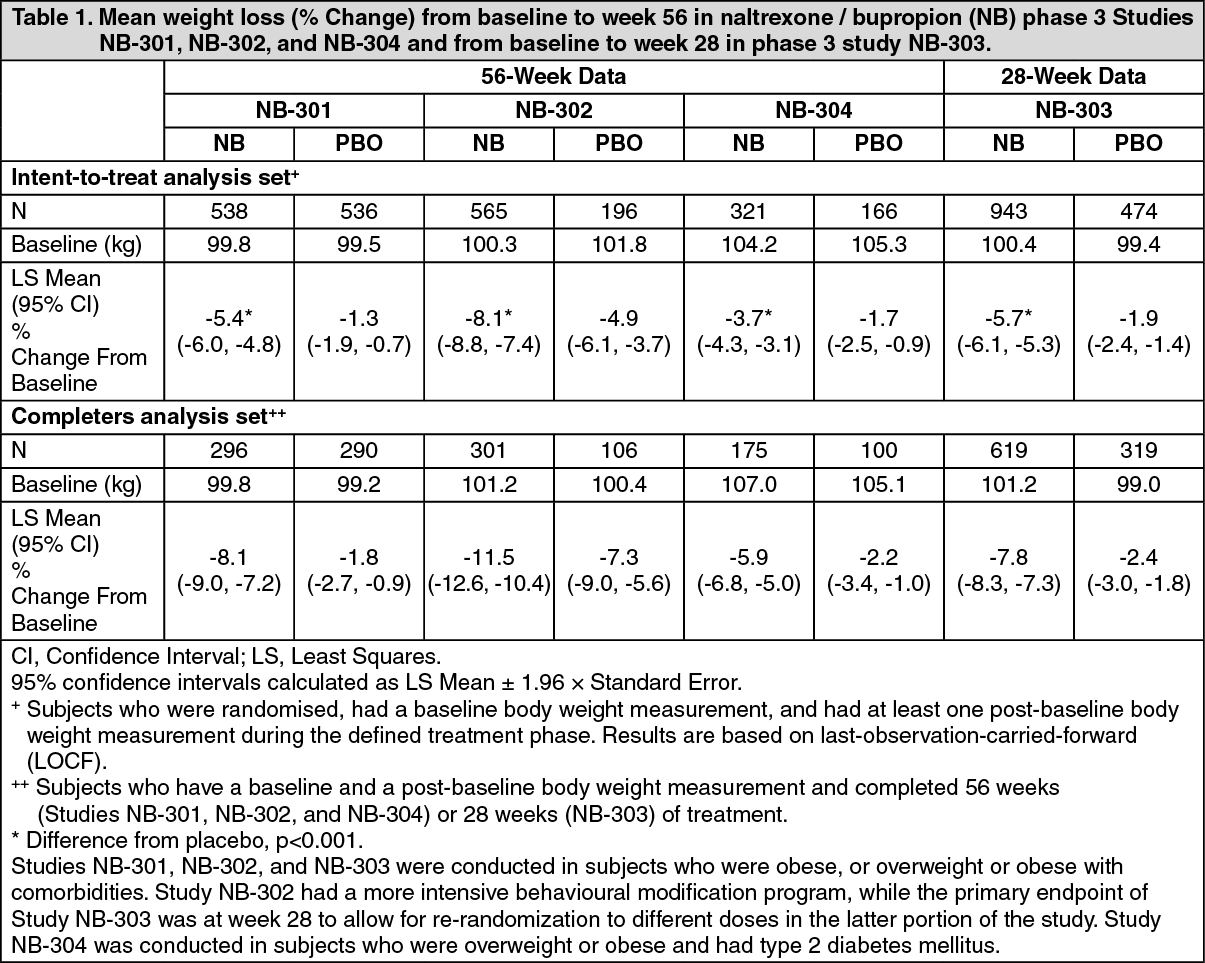
As seen in Table 1, in the NB-301 study subjects had a mean percent body weight loss of -5.4% while receiving naltrexone/bupropion compared to -1.3% in placebo-treated subjects. Weight loss of at least 5% baseline body weight was observed more frequently for subjects treated with naltrexone/bupropion (31%) compared to placebo (12%) (Table 1). More pronounced weight loss was observed in the cohort of subjects who completed 56 weeks of treatment with naltrexone/bupropion (-8.1%) compared to placebo (-1.8%). Comparable results were seen in the NB-303 study, which was of similar design, with significant weight loss observed in naltrexone/bupropion-treated subjects compared to placebo at the week 28 primary endpoint, and sustained through 56 weeks from baseline (Table 1).
Naltrexone/bupropion was also evaluated in combination with intensive behavioural modification counseling in the NB-302 study. Correspondingly, there was greater mean weight loss from baseline for naltrexone/bupropion treatment (-8.1%) compared to study NB-301 (-5.4%) at week 56, and for placebo (-4.9%) compared to study NB-301 (-1.3%).
The treatment effects observed in obese and overweight subjects with type 2 diabetes mellitus (Study NB-304) were somewhat less pronounced than those observed in the other Phase 3 studies. Naltrexone/bupropion (-3.7%) was significantly (p<0.001) more efficacious than placebo (-1.7%) treatment in this population. (See Table 1.)
 Click on icon to see table/diagram/image
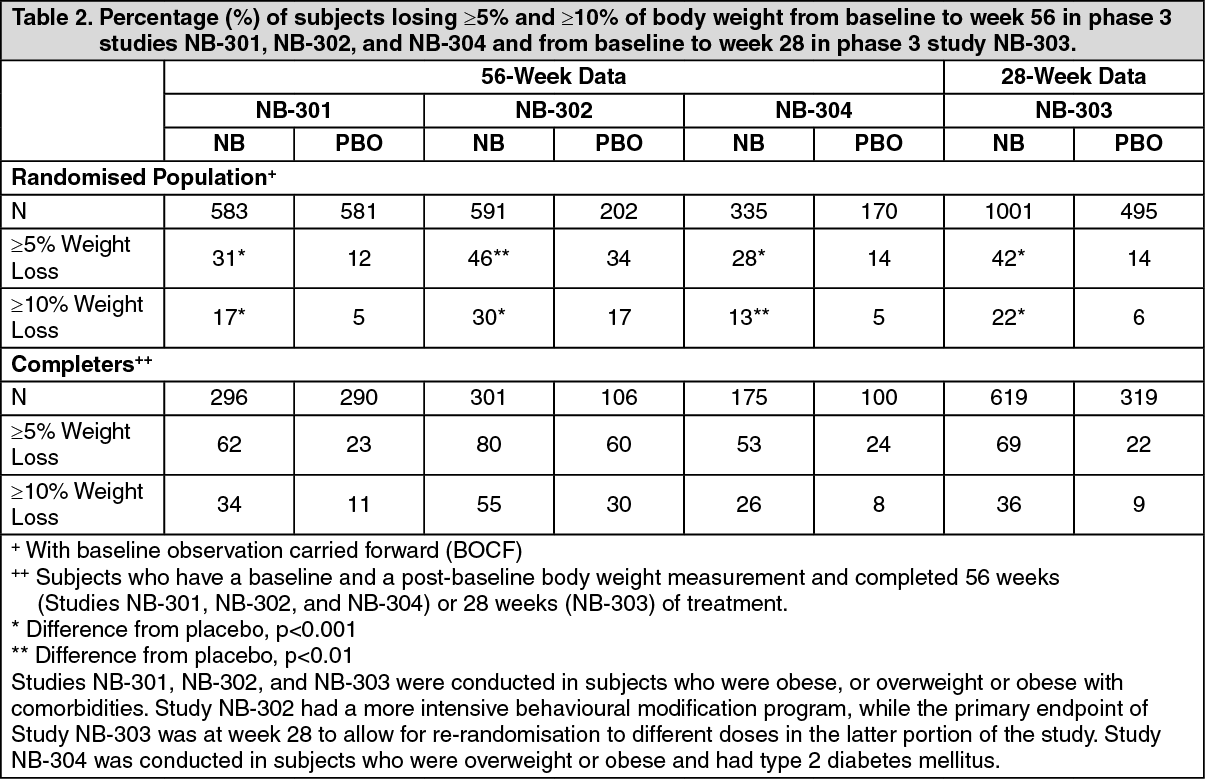
Click on icon to see table/diagram/imageThe percentages of subjects with ≥5% or ≥10% body weight loss from baseline were greater with naltrexone/bupropion compared to placebo in all four Phase 3 obesity trials (Table 2). (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageOf the subjects with observed data at week 16 in the four Phase 3 clinical trials, 50.8% of those randomised to receive naltrexone/bupropion had lost ≥5% of their baseline body weight, compared to 19.3% of placebo-treated subjects (week 16 Responders). At one year, the average weight loss (using LOCF methodology) among these week 16 Responders who received naltrexone/bupropion was 11.3%, with 55% losing ≥10% bodyweight. Additionally, week 16 Responders who received naltrexone/bupropion had a high retention rate with 87% completing 1 year of treatment. The ≥5% weight loss threshold at week 16 had 86.4% positive predictive value and 84.8% negative predictive value for determining whether a subject treated with naltrexone/bupropion would achieve at least 5% weight loss at week 56. Patients who did not meet the early response criterion were not found to have increased tolerability or safety issues relative to patients who did have a favourable early response.
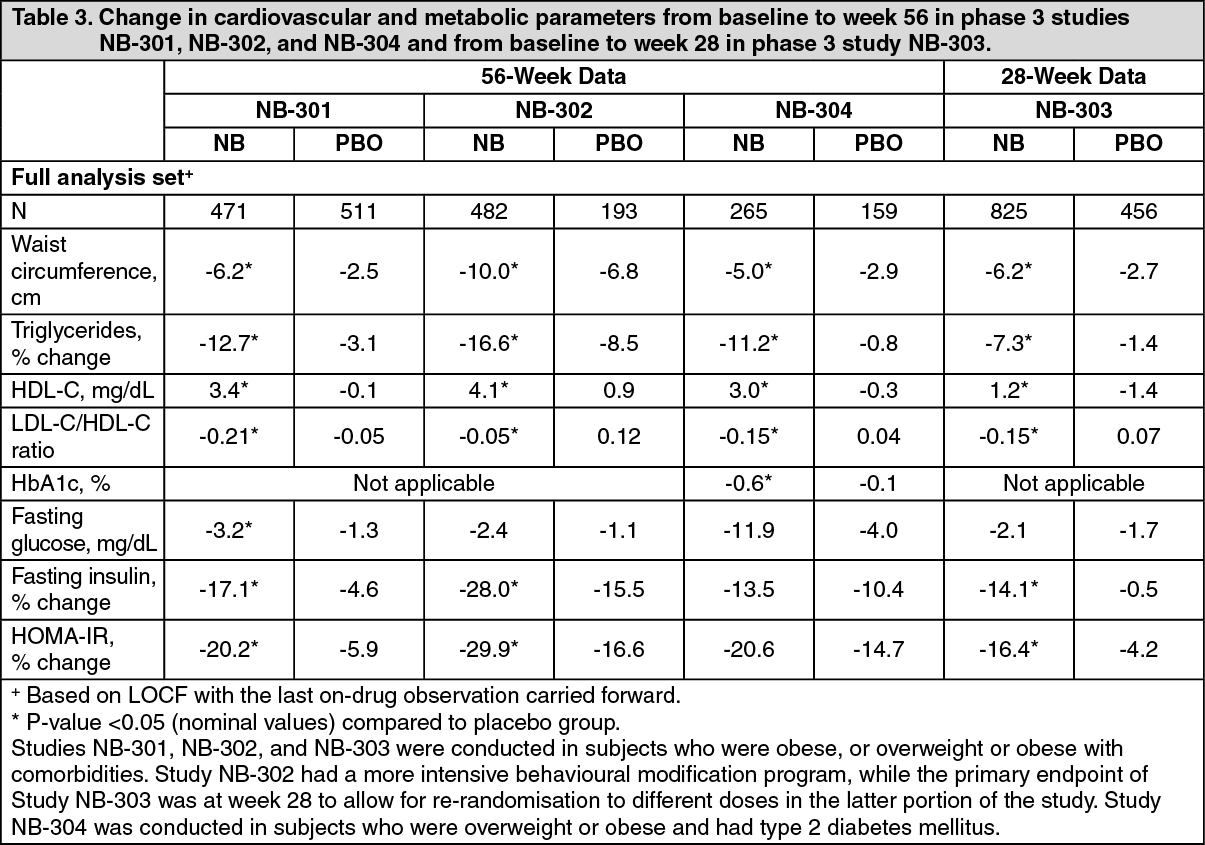
Effect on cardiovascular and metabolic parameters: Improvements were observed for waist circumference (including subjects with type 2 diabetes), triglycerides, HDL-C and LDL-C/HDL-C ratio for subjects treated with naltrexone/bupropion vs. placebo in all Phase 3 studies (Table 3). Improvements in triglycerides, HDL-C and LDL-C/HDL-C ratio were seen in naltrexone/bupropion-treated subjects diagnosed with baseline dyslipidaemia irrespective of dyslipidaemia treatment. Changes in mean blood pressure are described in Precautions. In addition, in subjects who did not have type 2 diabetes, there were reductions in fasting insulin and HOMA-IR, a measure of insulin resistance, in naltrexone/bupropion-treated subjects.
Effects on glycaemic control in obese subjects with type 2 diabetes: After 56 weeks of treatment in subjects with type 2 diabetes (NB-304), naltrexone/bupropion exhibited improvements in glycaemic control parameters compared to placebo (Table 3). Greater HbA1c improvement compared to placebo was observed at the first post-baseline measurement (week 16, p<0.001). Mean HbA1c change from baseline at week 56 was -0.63% for subjects treated with naltrexone/bupropion compared to subjects on placebo -0.14% (p<0.001). In subjects with baseline HbA1c >8% (64 mmol/mol), HbA1c changes at endpoint were -1.1% and -0.5% for naltrexone/bupropion compared to placebo, respectively. Improvements were observed for fasting glucose, fasting insulin, HOMA-IR and percent of subjects requiring rescue diabetes medicinal products for subjects treated with naltrexone/bupropion vs. placebo. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEffect on body composition: In a subset of subjects, body composition was measured using dual energy X-ray absorptiometry (DEXA) (naltrexone/bupropion = 79 subjects and placebo = 45 subjects) and multislice computed tomography (CT) scan (naltrexone/bupropion = 34 subjects and placebo = 24 subjects). The DEXA assessment showed that treatment with naltrexone/bupropion was associated with greater reductions from baseline in total body fat and in visceral adipose tissue than placebo. As expected, naltrexone/bupropion-treated subjects had a greater mean increase from baseline compared with placebo-treated subjects in percent of total body lean mass. These results suggest that most of the total weight loss was attributable to a reduction in adipose tissue, including visceral adipose.
Paediatric population: The European Medicines Agency has deferred the obligation to submit the results of studies with Contrave in one or more subsets of the paediatric population in obesity (see Dosage & Administration for information on paediatric use). Naltrexone/bupropion should not be used in children and adolescents.
Pharmacokinetics: The results of a single-dose relative bioavailability study in healthy subjects demonstrated that naltrexone/bupropion tablets, when dose adjusted, are bioequivalent based on AUC0-∞ mean ratio and 90% confidence intervals to naltrexone immediate-release (IR) or bupropion prolonged-release (PR) administered as single agents.
Absorption: Following single oral administration of naltrexone/bupropion tablets to healthy subjects, peak concentrations of naltrexone and bupropion occurred approximately 2 and 3 hours post administration of naltrexone/bupropion, respectively. There were no differences in bioavailability, as measured by AUC, of naltrexone or bupropion when administered in combination compared to each administered alone. However, given the prolonged nature of the drug release for naltrexone/bupropion, Cmax for naltrexone was markedly reduced compared to the 50 mg naltrexone hydrochloride IR administered alone (about 2-fold difference after dose adjustment). The bupropion Cmax from naltrexone/bupropion (180 mg bupropion hydrochloride) was equivalent to the Cmax of bupropion PR (150 mg bupropion hydrochloride), indicating that the bupropion Cmax achieved with naltrexone/bupropion (360 mg bupropion hydrochloride/day) is comparable to that achieved with commercially available bupropion PR (300 mg bupropion hydrochloride/day) administered alone.
Naltrexone and bupropion are well absorbed from the gastrointestinal tract (>90% absorbed), however, naltrexone has a significant first pass effect thereby limiting systemic bioavailability, with only 5-6% reaching the systemic circulation intact.
Food effect: When naltrexone/bupropion was given with a high-fat meal the AUC and Cmax for naltrexone increased 2.1-fold and 3.7-fold and the AUC and Cmax for bupropion increased 1.4-fold and 1.8-fold, respectively. At steady state, the food effect resulted in AUC and Cmax increases of 1.7- and 1.9-fold for naltrexone, and 1.1- and 1.3-fold for bupropion, respectively. Clinical experience included varying prandial conditions and supports the use of naltrexone/bupropion tablets with food.
Distribution: The mean volume of distribution at steady state of oral naltrexone and bupropion given as naltrexone/bupropion, Vss/F, was 5697 liters and 880 liters, respectively.
Plasma protein binding is not extensive for naltrexone (21%) or bupropion (84%) indicating low potential for drug interactions by displacement.
Biotransformation and elimination: Following single oral administration of naltrexone/bupropion tablets to healthy subjects, mean T½ elimination half-life was approximately 5 hours for naltrexone and 21 hours for bupropion.
Naltrexone: The major metabolite of naltrexone is 6-beta-naltrexol. Though less potent than naltrexone, 6-beta-naltrexol is eliminated more slowly and thus circulates at much higher concentrations than naltrexone. Naltrexone and 6-beta-naltrexol are not metabolised by cytochrome P450 enzymes and in vitro studies indicate that there is no potential for inhibition or induction of important isozymes. Naltrexone is primarily metabolised to 6-beta-naltrexol by the dihydrodiol dehydrogenases (DD1, DD2 and DD4). Other major metabolic routes are the formation of the metabolites 2-hydroxy-3-O-methyl naltrexone and 2-hydroxy-3-O-methyl-6-beta-naltrexol, believed to be mediated by catechol-O-methyl transferases (COMT), and glucuronidation, thought to be mediated by UGT1A1 and UGT2B7.
Naltrexone and its metabolites are excreted primarily by the kidney (37 to 60% of the dose). The derived value for renal excretion of naltrexone after oral administration, adjusting for plasma protein binding, is 89 mL/min. The enzyme responsible for the main elimination pathway is not known. Faecal excretion is a minor elimination pathway.
Bupropion: Bupropion is extensively metabolised with three active metabolites: hydroxybupropion, threohydrobupropion and erythrohydrobupropion. The metabolites have longer elimination half-lives than bupropion and accumulate to a greater extent. In vitro findings suggest that CYP2B6 is the principal isozyme involved in the formation of hydroxybupropion, while CYP1A2, 2A6, 2C9, 3A4 and 2E1 are less involved. In contrast, formation of threohydrobupropion has been reported in the literature to be mediated by 11-beta-hydroxysteroid dehydrogenase 1. The metabolic pathway responsible for the formation of erythrohydrobupropion is unknown.
Bupropion and its metabolites inhibit CYP2D6. Plasma protein binding of hydroxybupropion is similar to that of bupropion (84%) whereas the other two metabolites have approximately half the binding.
Following oral administration of 200 mg of 14C-bupropion hydrochloride in humans, 87% and 10% of the radioactive dose were recovered in the urine and feces, respectively. The fraction of the oral dose of bupropion excreted unchanged was 0.5%, a finding consistent with the extensive metabolism of bupropion.
Accumulation: Following twice daily administration of naltrexone/bupropion, naltrexone does not accumulate, while 6-beta-naltrexol accumulates over time. Based on its half-life, 6-beta-naltrexol is estimated to reach steady state concentrations in approximately 3 days. Metabolites of bupropion (and to a lesser extent unmetabolised bupropion) accumulate and reach steady state concentrations in approximately one week. No study has been performed comparing AUC or Cmax of naltrexone/bupropion prolonged-release tablets with bupropion PR or naltrexone IR administered as single agents in the multiple-dose setting (i.e., under steady state conditions).
Special populations: Gender and race: Pooled analysis of naltrexone/bupropion data revealed no meaningful gender- or race-related differences in the pharmacokinetic parameters of bupropion or naltrexone. However, only Caucasian and Black subjects were investigated to a significant extent. No dosage adjustment is necessary based on gender or race.
Elderly people: The pharmacokinetics of naltrexone/bupropion have not been evaluated in the elderly population. Because naltrexone and bupropion metabolic products are excreted in the urine and elderly people are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. Naltrexone/bupropion is not recommended in patients over 75 years of age.
Smokers: Pooled analysis of naltrexone/bupropion data revealed no meaningful differences in the plasma concentrations of bupropion or naltrexone in smokers compared to nonsmokers. The effects of cigarette smoking on the pharmacokinetics of bupropion were studied in 34 healthy male and female volunteers; 17 were chronic cigarette smokers and 17 were nonsmokers. Following oral administration of a single 150 mg dose of bupropion hydrochloride, there was no statistically significant difference in Cmax, half-life, Tmax, AUC, or clearance of bupropion or its active metabolites between smokers and nonsmokers.
Hepatic impairment: A single-dose pharmacokinetic study has been conducted with naltrexone/bupropion in patients with hepatic impairment. The results from this study demonstrated that in patients with mild hepatic impairment (Child-Pugh scores of 5-6 [Class A]), there was a modest increase in naltrexone concentrations, but concentrations of bupropion and most other metabolites were mostly comparable and no more than doubled to those in patients with normal hepatic function. In patients with moderate (Child-Pugh scores of 7-9 [Class B]) and severe (Child-Pugh scores of 10 or higher [Class C]) hepatic impairment, increases in the maximum concentration of naltrexone of ~6- and ~30-fold were observed for the moderate and severe patients respectively, while increases in bupropion were ~2-fold for both groups. Increases of ~2- and ~4-fold for the area under the curve for bupropion were observed for patients with moderate and severe impairment respectively. There were no consistent changes in naltrexone or bupropion metabolites related to varying degrees of hepatic impairment. Naltrexone/bupropion is contraindicated in patients with severe hepatic impairment (see Contraindications) and is not recommended in patients with moderate hepatic impairment (see Precautions). In patients with mild hepatic impairment, the maximum recommended daily dose for naltrexone/bupropion should be reduced (see Dosage & Administration).
Renal impairment: A single-dose pharmacokinetic study has been conducted for naltrexone/bupropion in subjects with mild, moderate, and severe renal impairment, compared with subjects with normal renal function. The results from this study demonstrated that the area under the curve for plasma naltrexone and metabolites and for plasma bupropion and metabolites was increased by less than two-fold in patients with moderate and severe renal impairment, and smaller increases were observed for patients with mild renal impairment. Based on these results, there are no dose adjustments recommended for patients with mild renal impairment. For patients with moderate or severe renal impairment, the maximum recommended daily dose for naltrexone/bupropion should be reduced (see Dosage & Administration). Naltrexone/bupropion is contraindicated in end-stage renal failure (see Contraindications).
Toxicology: Preclinical safety data: The effects of combined bupropion and naltrexone use have not been studied in animals.
Non-clinical data on individual components reveal no special hazard for humans based on conventional studies of safety, pharmacology, repeated dose toxicity, genotoxicity, and carcinogenic potential. Any effects in non-clinical studies were observed only at exposures considered sufficiently in excess of the maximum human exposure indicating little relevance to clinical use. However, there is some evidence on hepatotoxicity with increasing dose, since reversible increases of liver enzymes have been found in humans with therapeutic and higher doses (see Precautions and Adverse Reactions). Liver changes are seen in animal studies with bupropion but these reflect the action of a hepatic enzyme inducer. At recommended doses in humans, bupropion does not induce its own metabolism. This suggests that the hepatic findings in laboratory animals have only limited importance in the evaluation and risk assessment of bupropion.
Reproduction toxicity: Naltrexone (100 mg/kg/day, approximately 30 times the dose of naltrexone in naltrexone/bupropion on a mg/m2 basis) caused a significant increase in pseudo-pregnancy in the rat. A decrease in the pregnancy rate of mated female rats also occurred. There was no effect on male fertility at this dose level. The relevance of these observations to human fertility is not known.
Naltrexone has been shown to have an embryocidal effect in rats dosed with 100 mg/kg/day of naltrexone (30 times the naltrexone/bupropion dose) prior to and throughout gestation, and in rabbits treated with 60 mg/kg/day of naltrexone (36 times the naltrexone/bupropion dose) during the period of organogenesis.
A fertility study of bupropion in rats at doses up to 300 mg/kg/day, or 8 times the bupropion dose provided by naltrexone/bupropion revealed no evidence of impaired fertility.
Genotoxicity: Naltrexone was negative in the following in vitro genotoxicity studies: bacterial reverse mutation assay (Ames test), the heritable translocation assay, CHO cell sister chromatid exchange assay, and the mouse lymphoma gene mutation assay. Naltrexone was also negative in an in vivo mouse micronucleus assay. In contrast, naltrexone tested positive in the following assays: Drosophila recessive lethal frequency assay, non-specific DNA damage in repair tests with E. coli and WI-38 cells, and urinalysis for methylated histidine residues. The clinical relevance of these equivocal findings is unknown.
Genotoxicity data indicate that bupropion is a weak bacterial mutagen, but not a mammalian mutagen, and therefore is of no concern as a human genotoxic agent. Mouse and rat studies confirm the absence of carcinogenicity in these species.