Pharmacology: Pharmacodynamics: Mechanism of Action: CRESTOR is a selective and competitive inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts 3-hydroxy-3-methylglutaryl coenzyme A to mevalonate, a precursor of cholesterol.
In vivo studies in animals, and
in vitro studies in cultured animal and human cells have shown rosuvastatin to have a high uptake into, and selectivity for, action in the liver, the target organ for cholesterol lowering. In
in vivo and
in vitro studies, rosuvastatin produces its lipid-modifying effects in two ways. First, it increases the number of hepatic LDL receptors on the cell-surface to enhance uptake and catabolism of LDL. Second, rosuvastatin inhibits hepatic synthesis of VLDL, which reduces the total number of VLDL and LDL particles.
Pharmacokinetics: Absorption: In clinical pharmacology studies in man, peak plasma concentrations of rosuvastatin were reached 3 to 5 hours following oral dosing. Both C
max and AUC increased in approximate proportion to CRESTOR dose. The absolute bioavailability of rosuvastatin is approximately 20%.
Administration of CRESTOR with food did not affect the AUC of rosuvastatin.
The AUC of rosuvastatin does not differ following evening or morning drug administration.
Distribution: Mean volume of distribution at steady-state of rosuvastatin is approximately 134 liters. Rosuvastatin is 88% bound to plasma proteins, mostly albumin. This binding is reversible and independent of plasma concentrations.
Metabolism: Rosuvastatin is not extensively metabolized; approximately 10% of a radiolabeled dose is recovered as metabolite. The major metabolite is N-desmethyl rosuvastatin, which is formed principally by cytochrome P450 2C9, and
in vitro studies have demonstrated that N-desmethyl rosuvastatin has approximately one-sixth to one-half the HMG-CoA reductase inhibitory activity of the parent compound. Overall, greater than 90% of active plasma HMG-CoA reductase inhibitory activity is accounted for by the parent compound.
Excretion: Following oral administration, rosuvastatin and its metabolites are primarily excreted in the feces (90%). The elimination half-life (t
1/2) of rosuvastatin is approximately 19 hours.
After an intravenous dose, approximately 28% of total body clearance was via the renal route, and 72% by the hepatic route.
Race: A population pharmacokinetic analysis revealed no clinically relevant differences in pharmacokinetics among Caucasian, Hispanic, and Black or Afro-Caribbean groups. However, pharmacokinetic studies, including one conducted in the US, have demonstrated an approximate 2-fold elevation in median exposure (AUC and C
max) in Asian subjects when compared with a Caucasian control group.
Gender: There were no differences in plasma concentrations of rosuvastatin between men and women.
Geriatric: There were no differences in plasma concentrations of rosuvastatin between the nonelderly and elderly populations (age ≥ 65 years).
Renal Impairment: Mild to moderate renal impairment (CL
cr ≥ 30 mL/min/1.73 m
2) had no influence on plasma concentrations of rosuvastatin. However, plasma concentrations of rosuvastatin increased to a clinically significant extent (about 3-fold) in patients with severe renal impairment (CL
cr < 30 mL/min/1.73 m
2) not receiving hemodialysis compared with healthy subjects (CL
cr > 80 mL/min/1.73 m
2).
Hemodialysis: Steady-state plasma concentrations of rosuvastatin in patients on chronic hemodialysis were approximately 50% greater compared with healthy volunteer subjects with normal renal function.
Hepatic Impairment: In patients with chronic alcohol liver disease, plasma concentrations of rosuvastatin were modestly increased.
In patients with Child-Pugh A disease, C
max and AUC were increased by 60% and 5%, respectively, as compared with patients with normal liver function. In patients with Child-Pugh B disease, C
max and AUC were increased 100% and 21%, respectively, compared with patients with normal liver function.
Drug-Drug Interactions: Rosuvastatin clearance is not dependent on metabolism by cytochrome P450 3A4 to a clinically significant extent.
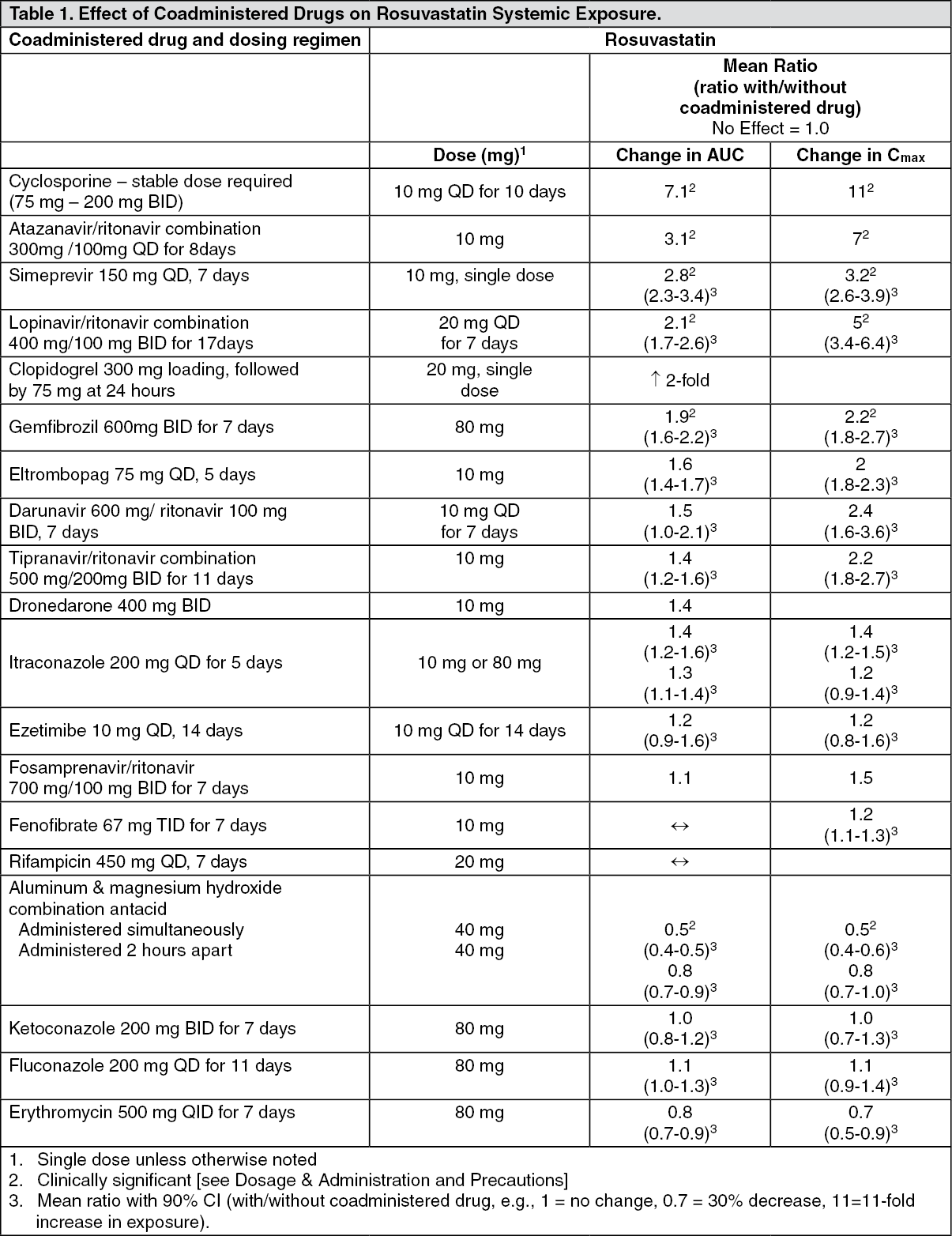
Rosuvastatin is a substrate for certain transporter proteins including the hepatic uptake transporter organic anion-transporting polyprotein 1B1 (OATP1B1) and efflux transporter breast cancer resistance protein (BCRP). Concomitant administration of CRESTOR with medications that are inhibitors of these transporter proteins (e.g. cyclosporine, certain HIV protease inhibitors) may result in increased rosuvastatin plasma concentrations and an increased risk of myopathy (see Dosage & Administration). It is recommended that prescribers consult the relevant product information when considering administration of such products together with CRESTOR. (See Tables 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pharmacogenomics:
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Pharmacogenomics: Disposition of HMG-CoA reductase inhibitors, including rosuvastatin, involves OATP1B1 and other transporter proteins. Higher plasma concentrations of rosuvastatin have been reported in very small groups of patients (n=3 to 5) who have two reduced function alleles of the gene that encodes OATP1B1 (
SLCO1B1 521T>C). The frequency of this genotype (i.e.,
SLCO1B1 521 C/C) is generally lower than 5% in most racial/ethnic groups. The impact of this polymorphism on efficacy and/or safety of rosuvastatin has not been clearly established. Doses should be titrated according to patient response and tolerability.
Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: In a 104-week carcinogenicity study in rats at dose levels of 2, 20, 60, or 80 mg/kg/day by oral gavage, the incidence of uterine stromal polyps was significantly increased in females at 80 mg/kg/day at systemic exposure 20 times the human exposure at 40 mg/day based on AUC. Increased incidence of polyps was not seen at lower doses.
In a 107-week carcinogenicity study in mice given 10, 60, 200 mg/kg/day by oral gavage, an increased incidence of hepatocellular adenoma/carcinoma was observed at 200 mg/kg/day at systemic exposures 20 times the human exposure at 40 mg/day based on AUC. An increased incidence of hepatocellular tumors was not seen at lower doses.
Rosuvastatin was not mutagenic or clastogenic with or without metabolic activation in the Ames test with
Salmonella typhimurium and
Escherichia coli, the mouse lymphoma assay, and the chromosomal aberration assay in Chinese hamster lung cells. Rosuvastatin was negative in the
in vivo mouse micronucleus test.
In rat fertility studies with oral gavage doses of 5, 15, 50 mg/kg/day, males were treated for 9 weeks prior to and throughout mating and females were treated 2 weeks prior to mating and throughout mating until gestation day 7. No adverse effect on fertility was observed at 50 mg/kg/day (systemic exposures up to 10 times the human exposure at 40 mg/day based on AUC). In testicles of dogs treated with rosuvastatin at 30 mg/kg/day for one month, spermatidic giant cells were seen. Spermatidic giant cells were observed in monkeys after 6-month treatment at 30 mg/kg/day in addition to vacuolation of seminiferous tubular epithelium. Exposures in the dog were 20 times and in the monkey 10 times the human exposure at 40 mg/day based on body surface area. Similar findings have been seen with other drugs in this class.
Animal Toxicology and/or Pharmacology: Embryo-fetal Development: Rosuvastatin crosses the placenta and is found in fetal tissue and amniotic fluid at 3% and 20%, respectively, of the maternal plasma concentration following a single 25 mg/kg oral gavage dose on gestation day 16 in rats. A higher fetal tissue distribution (25% maternal plasma concentration) was observed in rabbits after a single oral gavage dose of 1 mg/kg on gestation day 18.
In female rats given oral gavage doses of 5, 15, 50 mg/kg/day rosuvastatin before mating and continuing through day 7 postcoitus results in decreased fetal body weight (female pups) and delayed ossification at the high dose (systemic exposures 10 times the human exposure at 40 mg/day based on AUC).
In pregnant rats given oral gavage doses of 2, 10, 50 mg/kg/day from gestation day 7 through lactation day 21 (weaning), decreased pup survival occurred in groups given 50 mg/kg/day, systemic exposures ≥ 12 times the human exposure at 40 mg/day based on body surface area.
In pregnant rabbits given oral gavage doses of 0.3, 1, 3 mg/kg/day from gestation day 6 to lactation day 18 (weaning), exposures equivalent to the human exposure at 40 mg/day based on body surface area, decreased fetal viability and maternal mortality was observed.
Rosuvastatin was not teratogenic in rats at ≤ 25 mg/kg/day or in rabbits ≤ 3 mg/kg/day (systemic exposures equivalent to the human exposure at 40 mg/day based on AUC or body surface area, respectively).
Central Nervous System Toxicity: CNS vascular lesions, characterized by perivascular hemorrhages, edema, and mononuclear cell infiltration of perivascular spaces, have been observed in dogs treated with several other members of this drug class. A chemically similar drug in this class produced dose-dependent optic nerve degeneration (Wallerian degeneration of retinogeniculate fibers) in dogs, at a dose that produced plasma drug levels about 30 times higher than the mean drug level in humans taking the highest recommended dose. Edema, hemorrhage, and partial necrosis in the interstitium of the choroid plexus was observed in a female dog sacrificed moribund at day 24 at 90 mg/kg/day by oral gavage (systemic exposures 100 times the human exposure at 40 mg/day based on AUC). Corneal opacity was seen in dogs treated for 52 weeks at 6 mg/kg/day by oral gavage (systemic exposures 20 times the human exposure at 40 mg/day based on AUC). Cataracts were seen in dogs treated for 12 weeks by oral gavage at 30 mg/kg/day (systemic exposures 60 times the human exposure at 40 mg/day based on AUC). Retinal dysplasia and retinal loss were seen in dogs treated for 4 weeks by oral gavage at 90 mg/kg/day (systemic exposures 100 times the human exposure at 40 mg/day based on AUC). Doses ≤ 30 mg/kg/day (systemic exposures ≤ 60 times the human exposure at 40 mg/day based on AUC) did not reveal retinal findings during treatment for up to one year.
Clinical Studies: Hyperlipidemia and Mixed Dyslipidemia: CRESTOR reduces Total-C, LDL-C, ApoB, nonHDL-C, and TG, and increases HDL-C, in adult patients with hyperlipidemia and mixed dyslipidemia.
Dose-Ranging Study: In a multicenter, double-blind, placebo-controlled, dose-ranging study in patients with hyperlipidemia CRESTOR given as a single daily dose for 6 weeks significantly reduced Total -C, LDL-C, nonHDL-C, and ApoB, across the dose range (Table 3).
Click on icon to see table/diagram/image
Active-Controlled Study: CRESTOR was compared with the HMG-CoA reductase inhibitors atorvastatin, simvastatin, and pravastatin in a multicenter, open-label, dose-ranging study of 2,240 patients with hyperlipidemia or mixed dyslipidemia. After randomization, patients were treated for 6 weeks with a single daily dose of either CRESTOR, atorvastatin, simvastatin, or pravastatin (Figure 1 and Table 4).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Heterozygous Familial Hypercholesterolemia: Active-Controlled Study: In a study of patients with heterozygous FH (baseline mean LDL of 291), patients were randomized to CRESTOR 20 mg or atorvastatin 20 mg. The dose was increased by 6-week intervals. Significant LDL-C reductions from baseline were seen at each dose in both treatment groups (Table 5).
Click on icon to see table/diagram/image
Hypertriglyceridemia: Dose-Response Study: In a double-blind, placebo-controlled dose-response study in patients with baseline TG levels from 273 to 817 mg/dL, CRESTOR given as a single daily dose (5 to 40 mg) over 6 weeks significantly reduced serum TG levels (Table 6).
Click on icon to see table/diagram/image
Primary Dysbetalipoproteinemia (Type III Hyperlipoproteinemia): In a randomized, multicenter, double-blind crossover study, 32 patients (27 with ε2/ε2 and 4 with apo E mutation [Arg145Cys] with primary dysbetalipoproteinemia (Type III Hyperlipoproteinemia) entered a 6-week dietary lead-in period on the NCEP Therapeutic Lifestyle Change (TLC) diet. Following dietary lead-in, patients were randomized to a sequence of treatments in conjunction with the TLC diet for 6 weeks each: rosuvastatin 10 mg followed by rosuvastatin 20 mg or rosuvastatin 20 mg followed by rosuvastatin 10 mg. CRESTOR reduced nonHDL-C (primary end point) and circulating remnant lipoprotein levels. Results are shown in the table as follows. (See Table 7.)
Click on icon to see table/diagram/image
Homozygous Familial Hypercholesterolemia: Dose-Titration Study: In an open-label, forced-titration study, homozygous FH patients (n=40, 8-63 years) were evaluated for their response to CRESTOR 20 to 40 mg titrated at a 6-week interval. In the overall population, the mean LDL-C reduction from baseline was 22%. About one-third of the patients benefited from increasing their dose from 20 mg to 40 mg with further LDL lowering of greater than 6%. In the 27 patients with at least a 15% reduction in LDL-C, the mean LDL-C reduction was 30% (median 28% reduction). Among 13 patients with an LDL-C reduction of <15%, 3 had no change or an increase in LDL-C. Reductions in LDL-C of 15% or greater were observed in 3 of 5 patients with known receptor negative status.
Pediatric Patients with Heterozygous Familial Hypercholesterolemia: In a double-blind, randomized, multicenter, placebo-controlled, 12-week study, 176 (97 male and 79 female) children and adolescents with heterozygous familial hypercholesterolemia were randomized to rosuvastatin 5, 10, or 20 mg or placebo daily. Patients ranged in age from 10 to 17 years (median age of 14 years) with approximately 30% of the patients 10 to 13 years and approximately 17%, 18%, 40%, and 25% at Tanner stages II, III, IV, and V, respectively. Females were at least 1 year postmenarche. Mean LDL-C at baseline was 233 mg/dL (range of 129 to 399). The 12-week double-blind phase was followed by a 40-week open-label dose-titration phase, where all patients (n=173) received 5 mg, 10 mg, or 20 mg rosuvastatin daily.
Rosuvastatin significantly reduced LDL-C (primary end point), total cholesterol and ApoB levels at each dose compared to placebo. Results are shown in Table 8 as follows. (See Table 8.)
Click on icon to see table/diagram/image
At the end of the 12-week, double-blind treatment period, the percentage of patients achieving the LDL-C goal of less than 110 mg/dL (2.8 mmol/L) was 0% for placebo, 12% for rosuvastatin 5 mg, 41% for rosuvastatin 10 mg and 41% for rosuvastatin 20 mg. For the 40-week, open-label phase, 71% of the patients were titrated to the maximum dose of 20 mg and 41% of the patients achieved the LDL-C goal of 110 mg/dL.
The long-term efficacy of rosuvastatin therapy initiated in childhood to reduce morbidity and mortality in adulthood has not been established.
Slowing of the Progression of Atherosclerosis: In the Measuring Effects on Intima Media Thickness: an Evaluation Of Rosuvastatin 40 mg (METEOR) study, the effect of therapy with CRESTOR on carotid atherosclerosis was assessed by B-mode ultrasonography in patients with elevated LDL-C, at low risk (Framingham risk <10% over ten years) for symptomatic coronary artery disease and with subclinical atherosclerosis as evidenced by carotid intimal-medial thickness (cIMT). In this double-blind, placebo-controlled clinical study 984 patients were randomized (of whom 876 were analyzed) in a 5:2 ratio to CRESTOR 40 mg or placebo once daily. Ultrasonograms of the carotid walls were used to determine the annualized rate of change per patient from baseline to two years in mean maximum cIMT of 12 measured segments. The estimated difference in the rate of change in the maximum cIMT analyzed over all 12 carotid artery sites between patients treated with CRESTOR and placebo-treated patients was -0.0145 mm/year (95% CI -0.0196, -0.0093; p<0.0001).
The annualized rate of change from baseline for the placebo group was +0.0131 mm/year (p<0.0001). The annualized rate of change from baseline for the group treated with CRESTOR was -0.0014 mm/year (p=0.32).
At an individual patient level in the group treated with CRESTOR, 52.1% of patients demonstrated an absence of disease progression (defined as a negative annualized rate of change), compared to 37.7% of patients in the placebo group.
Primary Prevention of Cardiovascular Disease: In the Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) study, the effect of CRESTOR (rosuvastatin calcium) on the occurrence of major cardiovascular (CV) disease events was assessed in 17,802 men (≥50 years) and women (≥60 years) who had no clinically evident cardiovascular disease, LDL-C levels <130 mg/dL (3.3 mmol/l) and hs-CRP levels ≥2 mg/L. The study population had an estimated baseline coronary heart disease risk of 11.6% over 10 years based on the Framingham risk criteria and included a high percentage of patients with additional risk factors such as hypertension (58%), low HDL-C levels (23%), cigarette smoking (16%), or a family history of premature CHD (12%). Study participants had a median baseline LDL-C of 108 mg/dL and hsCRP of 4.3 mg/L. Study participants were randomly assigned to placebo (n=8901) or rosuvastatin 20 mg once daily (n=8901) and were followed for a mean duration of 2 years. The JUPITER study was stopped early by the Data Safety Monitoring Board due to meeting predefined stopping rules for efficacy in rosuvastatin-treated subjects.
The primary end point was a composite end point consisting of the time-to-first occurrence of any of the following major CV events: CV death, nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina or an arterial revascularization procedure.
Rosuvastatin significantly reduced the risk of major CV events (252 events in the placebo group vs. 142 events in the rosuvastatin group) with a statistically significant (p<0.001) relative risk reduction of 44% and absolute risk reduction of 1.2% (see Figure 2). The risk reduction for the primary end point was consistent across the following predefined subgroups: age, sex, race, smoking status, family history of premature CHD, body mass index, LDL-C, HDL-C, and hsCRP levels. (See Figure 2.)
Click on icon to see table/diagram/image
The individual components of the primary end point are presented in Figure 3. Rosuvastatin significantly reduced the risk of nonfatal myocardial infarction, nonfatal stroke, and arterial revascularization procedures. There were no significant treatment differences between the rosuvastatin and placebo groups for death due to cardiovascular causes or hospitalizations for unstable angina.
Rosuvastatin significantly reduced the risk of myocardial infarction (6 fatal events and 62 nonfatal events in placebo-treated subjects vs. 9 fatal events and 22 nonfatal events in rosuvastatin treated subjects) and the risk of stroke (6 fatal events and 58 nonfatal events in placebo-treated subjects vs. 3 fatal events and 30 nonfatal events in rosuvastatin-treated subjects).
In a post-hoc subgroup analysis of JUPITER subjects (n=1405; rosuvastatin=725, placebo=680) with a hsCRP ≥2 mg/L and no other traditional risk factors (smoking, BP ≥140/90 or taking antihypertensives, low HDL-C) other than age, after adjustment for high HDL-C, there was no significant treatment benefit with rosuvastatin treatment. (See Figure 3.)
Click on icon to see table/diagram/image
At one year, rosuvastatin increased HDL-C and reduced LDL-C, hsCRP, total cholesterol and serum triglyceride levels (p<0.001 for all versus placebo).





 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image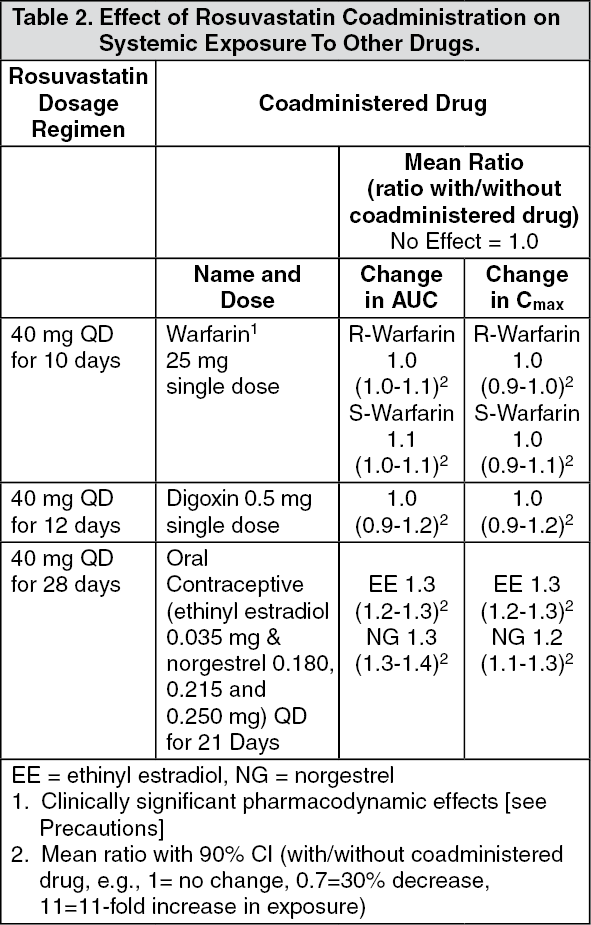 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image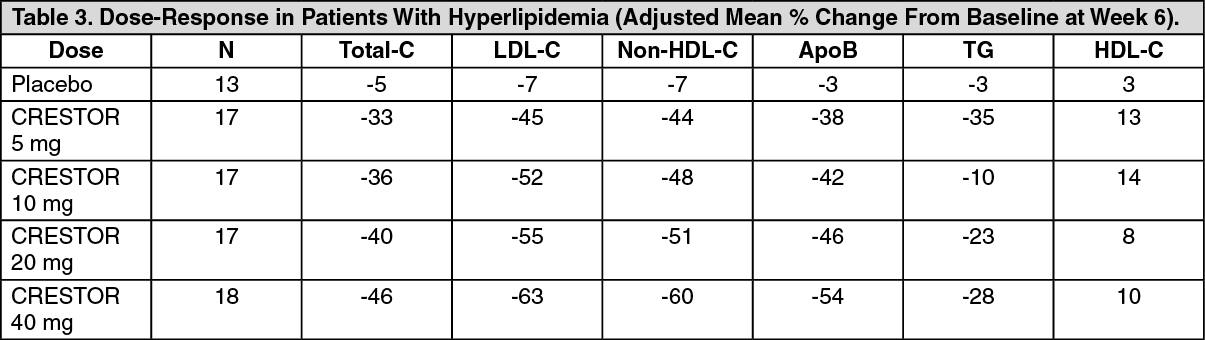 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image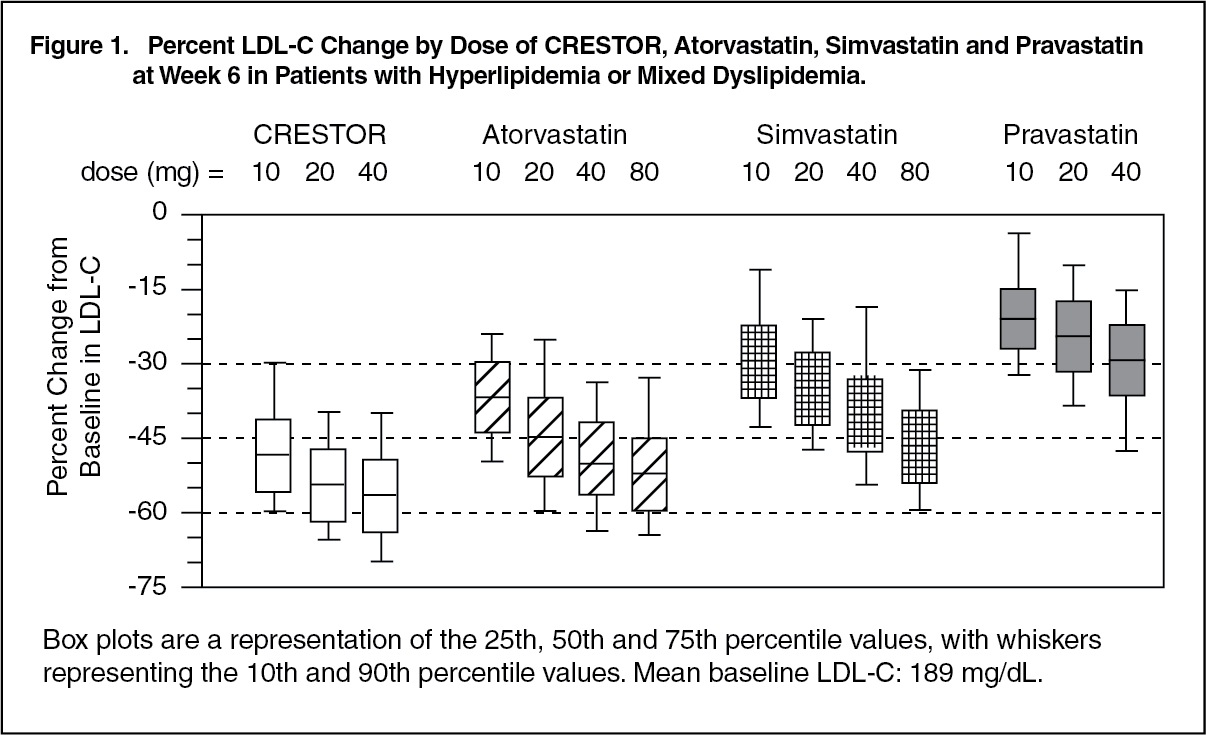 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image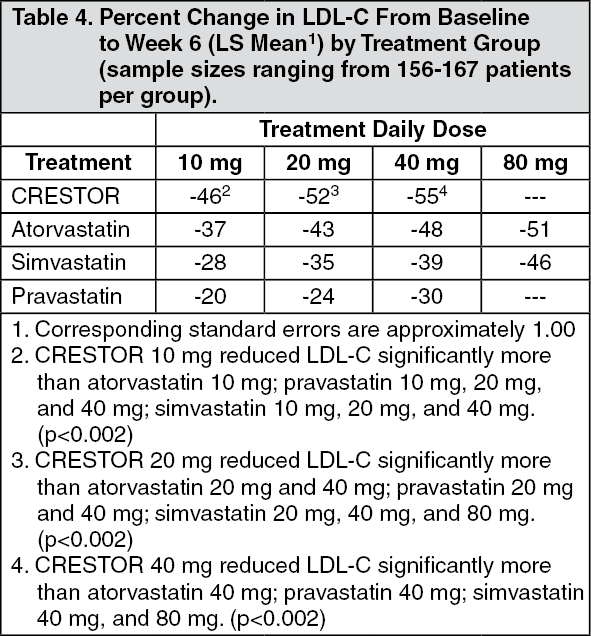 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image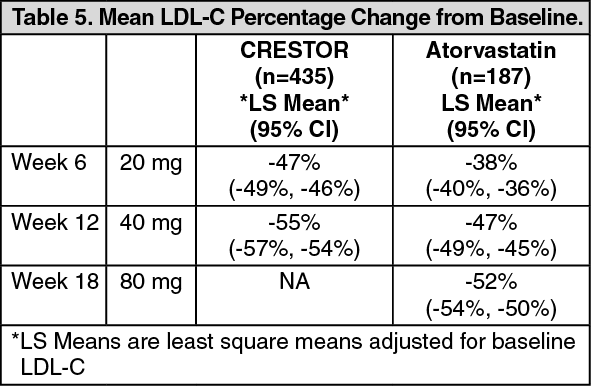 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image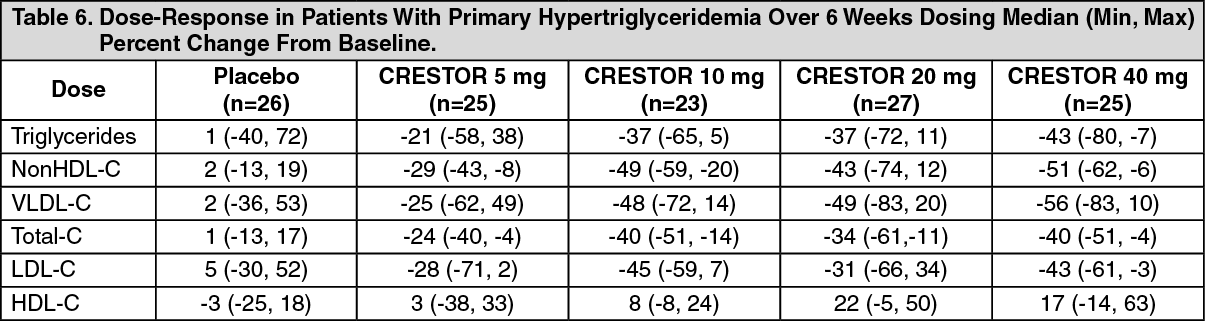 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image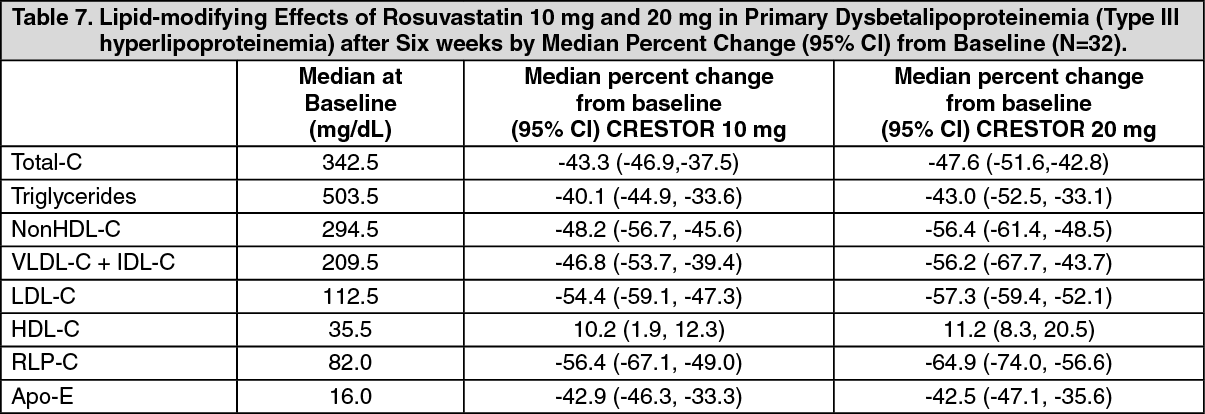 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image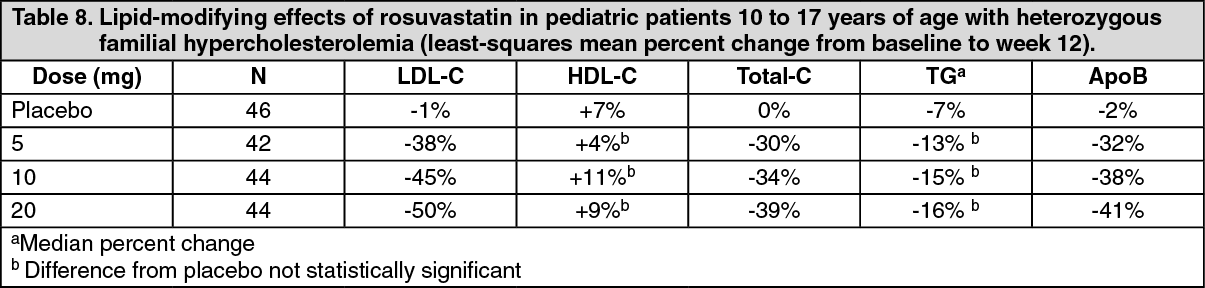 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image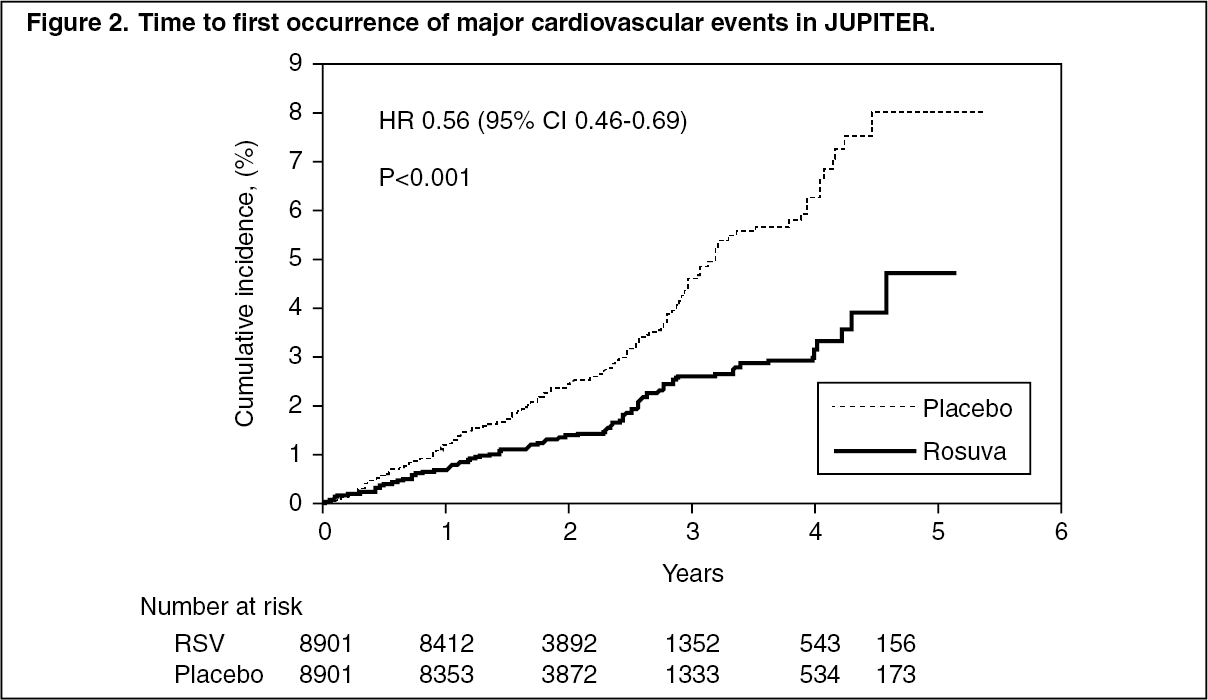 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image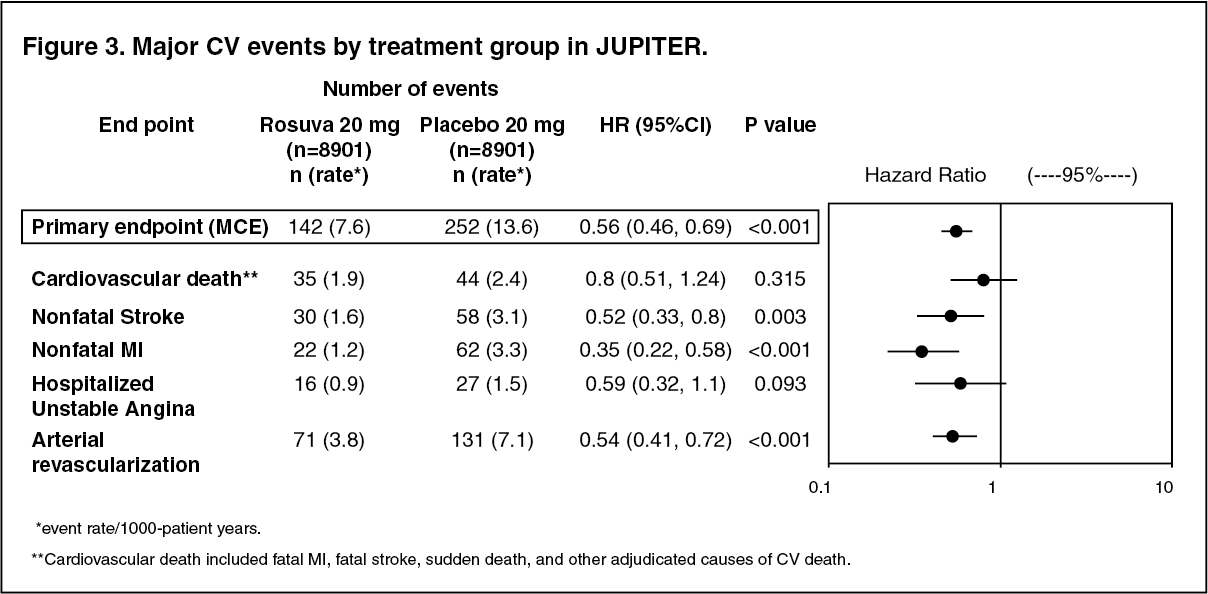 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image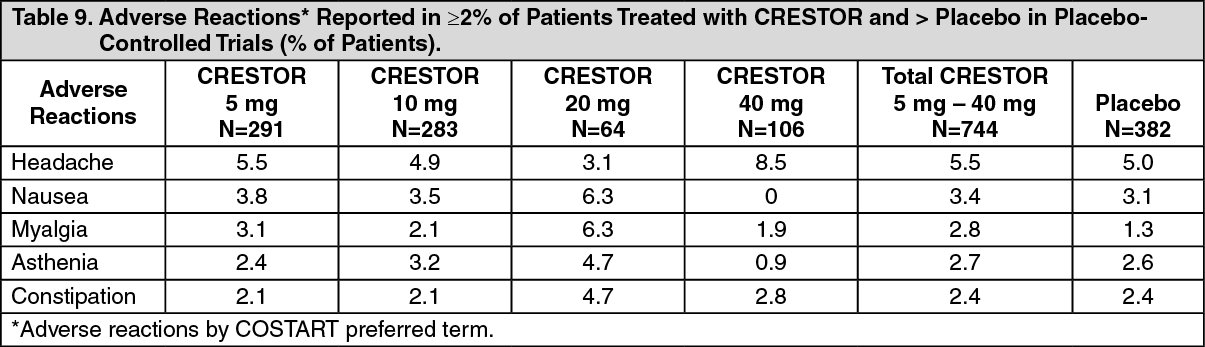 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image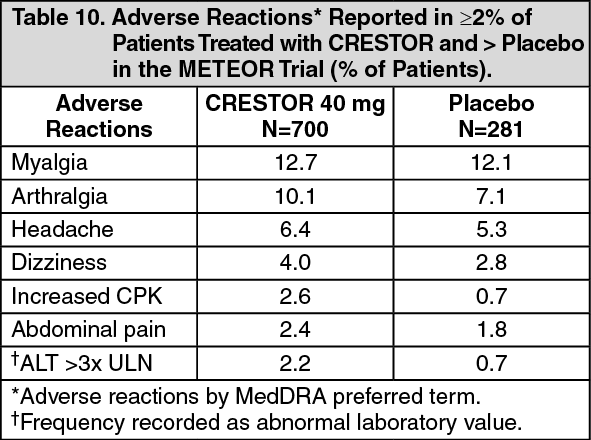 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image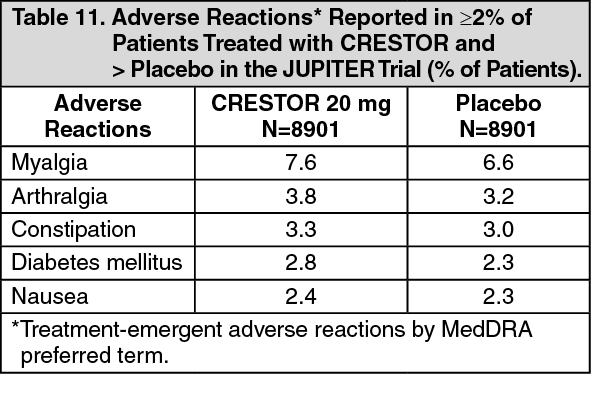 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image



 Sign Out
Sign Out