Pharmacotherapeutic group: Drugs for obstructive airway diseases, other systemic drugs for obstructive airway diseases.
ATC code: R03DX07.
Pharmacology: Pharmacodynamics: Mechanism of action: Roflumilast is a PDE4 inhibitor, a non-steroid, anti-inflammatory active substance designed to target both the systemic and pulmonary inflammation associated with COPD. The mechanism of action is the inhibition of PDE4, a major cyclic adenosine monophosphate (cAMP)-metabolizing enzyme found in structural and inflammatory cells important to the pathogenesis of COPD. Roflumilast targets the PDE4A, 4B and 4D splicing variants with similar potency in the nanomolar range. The affinity to the PDE4C splicing variants is 5 to 10-fold lower. This mechanism of action and the selectivity also apply to roflumilast N-oxide, which is the major active metabolite of roflumilast.
Pharmacodynamic effects: Inhibition of PDE4 leads to elevated intracellular cAMP levels and mitigates COPD-related malfunctions of leukocytes, airway and pulmonary vascular smooth muscle cells, endothelial and airway epithelial cells and fibroblasts in experimental models. Upon
in vitro stimulation of human neutrophils, monocytes, macrophages or lymphocytes, roflumilast and roflumilast N-oxide suppress the release of inflammatory mediators e.g. leukotriene B4, reactive oxygen species, tumor necrosis factor α, interferon γ and granzyme B.
In patients with COPD, roflumilast reduced sputum neutrophils. Furthermore, roflumilast attenuated influx of neutrophils and eosinophils into the airways of endotoxin challenged healthy volunteers.
Clinical efficacy and safety: In two confirmative replicate one-year studies (M2-124 and M2-125) and two supplementary six-month studies (M2-127 and M2-128), a total number of 4,768 patients were randomized and treated of whom 2,374 were treated with roflumilast. The design of the studies was parallel-group, double-blind and placebo-controlled.
The one-year studies included patients with a history of severe to very severe COPD [FEV
1 (forced expiratory volume in one second) ≤50% of predicted] associated with chronic bronchitis, with at least one documented exacerbation in the previous year and with symptoms at baseline as determined by cough and sputum score. Long-acting beta-agonists (LABAs) were allowed in the studies and were used in approximately 50% of the study population. Short-acting anticholinergics (SAMAs) were allowed for those patients not taking LABAs. Rescue medicinal products (salbutamol or albuterol) were allowed on an as-needed basis. The use of inhaled corticosteroids and theophylline was prohibited during the studies. Patients with no history of exacerbations were excluded.
In a pooled analysis of the one-year studies M2-124 and M2-125, roflumilast 500 micrograms once daily significantly improved lung function compared to placebo, on average by 48 ml (pre-bronchodilator FEV
1, primary endpoint, p<0.0001), and by 55 ml (post-bronchodilator FEV
1, p<0.0001). The improvement in lung function was apparent at the first visit after 4 weeks and was maintained up to one year (end of treatment period). The rate (per patient per year) of moderate exacerbations (requiring intervention with systemic glucocorticosteroids) or severe exacerbations (resulting in hospitalisation and/or leading to death) after 1 year was 1.142 with roflumilast and 1.374 with placebo corresponding to a relative risk reduction of 16.9% (95%CI: 8.2% to 24.8%) (primary endpoint, p=0.0003). Effects were similar, independent of previous treatment with inhaled corticosteroids or underlying treatment with LABAs. In the subgroup of patients with history of frequent exacerbations (at least 2 exacerbations during the last year), the rate of exacerbations was 1.526 with roflumilast and 1.941 with placebo corresponding to a relative risk reduction of 21.3% (95%CI: 7.5% to 33.1%). Roflumilast did not significantly reduce the rate of exacerbations compared with placebo in the subgroup of patients with moderate COPD.
The reduction of moderate or severe exacerbations with roflumilast and LABA compared to placebo and LABA was on average 21% (p=0.0011). The respective reduction in exacerbations seen in patients without concomitant LABAs was on average 15% (p=0.0387). The numbers of patients who died due to any reason were equal for those treated with placebo or roflumilast (42 deaths each group; 2.7% each group; pooled analysis).
A total of 2,690 patients were included and randomized in two supportive 1-year studies (M2-111 and M2-112). In contrast to the two confirmative studies, a history of chronic bronchitis and of COPD exacerbations was not requested for patients' inclusion. Inhaled corticosteroids were used in 809 (61%) of the roflumilast treated patients, whereas the use of LABAs and theophylline was prohibited. Roflumilast 500 micrograms once daily significantly improved lung function compared to placebo, on average by 51 ml (pre-bronchodilator FEV
1, p<0.0001), and by 53 ml (post-bronchodilator FEV
1, p<0.0001). The rate of exacerbations (as defined in the protocols) were not significantly reduced by roflumilast in the individual studies (relative risk reduction: 13.5% in study M2-111 and 6.6% in study M2-112; p=not significant). Adverse events rates were independent of concomitant treatment with inhaled corticosteroids.
Two six-month supportive studies (M2-127 and M2-128) included patients with a history of COPD for at least 12 months prior to baseline. Both studies included moderate to severe patients with a non-reversible airway obstruction and a FEV
1 of 40% to 70% of predicted. Roflumilast or placebo treatment was added to continuous treatment with a long-acting bronchodilator, in particular salmeterol in study M2-127 or tiotropium in study M2-128. In the two six-month studies, pre-bronchodilator FEV
1 was significantly improved by 49 ml (primary endpoint, p<0.0001) beyond the bronchodilator effect of concomitant treatment with salmeterol in study M2-127 and by 80 ml (primary endpoint, p<0.0001) incremental to concomitant treatment with tiotropium in study M2-128.
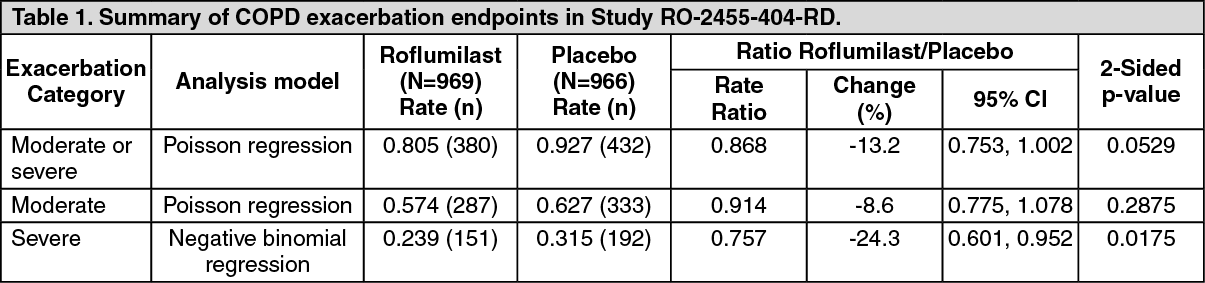
Study RO-2455-404-RD was a one year study in COPD patients with a baseline (pre-bronchodilator) FEV
1 <50% of predicted normal and a history of frequent exacerbations. The study assessed the effect of roflumilast on COPD exacerbation rate in patients treated with fixed combinations of LABA and inhaled corticosteroids, compared to placebo. A total of 1935 patients were randomised to double-blind medication and approximately 70% were also using a long-acting muscarinic antagonist (LAMA) through the course of the trial. The primary endpoint was reduction in rate of moderate or severe COPD exacerbations per patient per year. The rate of severe COPD exacerbations and changes in FEV
1 were evaluated as key secondary endpoints.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
There was a trend towards a reduction in moderate or severe exacerbations in subjects treated with roflumilast compared with placebo over 52 weeks, which did not achieve statistical significance (see Table 1). A pre-specified sensitivity analysis using the negative binomial regression model treatment showed a statistically significant difference of -14.2% (rate ratio: 0.86; 95% CI: 0.74 to 0.99).
The per-protocol Poisson regression analysis and the non-significant sensitivity to drop-out Poisson regression intention-to-treat analysis rate ratios were 0.81 (95% CI: 0.69 to 0.94) and 0.89 (95% CI: 0.77 to 1.02), respectively.
Reductions were achieved in the subgroup of patients concomitantly treated with LAMA (rate ratio: 0.88; 95% CI: 0.75 to 1.04) and in the subgroup not treated with LAMA (rate ratio: 0.83; 95% CI: 0.62 to 1.12).
The rate of severe exacerbations was reduced in the overall patient group (rate ratio: 0.76; 95% CI: 0.60 to 0.95) with a rate of 0.24 per patient/year compared to a rate of 0.32 per patient/year in patients treated with placebo. A similar reduction was achieved in the subgroup of patients concomitantly treated with LAMA (rate ratio: 0.77; 95% CI: 0.60 to 0.99) and in the subgroup not treated with LAMA (rate ratio: 0.71; 95% CI: 0.42 to 1.20).
Roflumilast improved lung function after 4 weeks (sustained over 52 weeks). Post-bronchodilator FEV
1 increased for the roflumilast group by 52 mL (95% CI: 40, 65 mL) and decreased for the placebo group by 4 mL (95% CI: -16, 9 mL). Post-bronchodilator FEV
1 showed a clinically significant improvement in favour of Roflumilast by 56 mL over placebo (95% CI: 38, 73 mL).
Seventeen (1.8%) patients in the roflumilast group and 18 (1.9%) patients in the placebo group died during the double-blind treatment period due to any reason and 7 (0.7%) patients in each group due to a COPD exacerbation. The proportion of patients who experienced at least 1 adverse event during the double-blind treatment period were 648 (66.9%) patients and 572 (59.2%) patients in the roflumilast and placebo groups, respectively. The observed adverse reactions for roflumilast in Study RO-2455-404-RD were in line with those already included in Adverse Reactions.
More patients in the roflumilast group (27.6%) than placebo (19.8%) withdrew study medication due to any reason (risk ratio: 1.40; 95%CI: 1.19 to 1.65). The major reasons for trial discontinuation were withdrawal of consent and reported adverse events.
Paediatric population: The European Medicines Agency has waived the obligation to submit the results of studies with roflumilast in all subsets of the paediatric population in chronic obstructive pulmonary disease (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: Roflumilast is extensively metabolised in humans, with the formation of a major pharmacodynamically active metabolite, roflumilast N-oxide. Since both roflumilast and roflumilast N-oxide contribute to PDE4 inhibitory activity
in vivo, pharmacokinetic considerations are based on total PDE4 inhibitory activity (i.e. total exposure to roflumilast and roflumilast N-oxide).
Absorption: The absolute bioavailability of roflumilast following a 500 micrograms oral dose is approximately 80%. Maximum plasma concentrations of roflumilast typically occur approximately one hour after dosing (ranging from 0.5 to 2 hours) in the fasted state. Maximum concentrations of the N-oxide metabolite are reached after about eight hours (ranging from 4 to 13 hours). Food intake does not affect the total PDE4 inhibitory activity, but delays time to maximum concentration (t
max) of roflumilast by one hour and reduces C
max by approximately 40%. However, C
max and t
max of roflumilast N-oxide are unaffected.
Distribution: Plasma protein binding of roflumilast and its N-oxide metabolite is approximately 99% and 97%, respectively. Volume of distribution for single dose of 500 micrograms roflumilast is about 2.9 l/kg. Due to the physico-chemical properties, roflumilast is readily distributed to organs and tissues including fatty tissue of mice, hamster and rat. An early distribution phase with marked penetration into tissues is followed by a marked elimination phase out of fatty tissue most probably due to pronounced break-down of parent compound to roflumilast N-oxide. These studies in rats with radiolabeled roflumilast also indicate low penetration across the blood-brain barrier. There is no evidence for a specific accumulation or retention of roflumilast or its metabolites in organs and fatty tissue.
Biotransformation: Roflumilast is extensively metabolised via Phase I (cytochrome P450) and Phase II (conjugation) reactions. The N-oxide metabolite is the major metabolite observed in the plasma of humans. The plasma AUC of the N-oxide metabolite on average is about 10-fold greater than the plasma AUC of roflumilast. Thus, the N-oxide metabolite is considered to be the main contributor to the total PDE4 inhibitory activity
in vivo.
In vitro studies and clinical interaction studies suggest that the metabolism of roflumilast to its N-oxide metabolite is mediated by CYP1A2 and 3A4. Based on further
in vitro results in human hepatic microsomes, therapeutic plasma concentrations of roflumilast and roflumilast N-oxide do not inhibit CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5, or 4A9/11. Therefore, there is a low probability of relevant interactions with substances metabolised by these P450 enzymes. In addition,
in vitro studies demonstrated no induction of the CYP1A2, 2A6, 2C9, 2C19, or 3A4/5 and only a weak induction of CYP2B6 by roflumilast.
Elimination: The plasma clearance after short-term intravenous infusion of roflumilast is about 9.6 l/h. Following an oral dose, the median plasma effective half-life of roflumilast and its N-oxide metabolite are approximately 17 and 30 hours, respectively. Steady state plasma concentrations of roflumilast and its N-oxide metabolite are reached after approximately 4 days for roflumilast and 6 days for roflumilast N-oxide following once-daily dosing. Following intravenous or oral administration of radiolabeled roflumilast, about 20% of the radioactivity was recovered in the faeces and 70% in urine as inactive metabolites.
Linearity/Non-linearity: The pharmacokinetics of roflumilast and its N-oxide metabolite are dose-proportional over a range of doses from 250 micrograms to 1,000 micrograms.
Special populations: In older people, females and in non-Caucasians, total PDE4 inhibitory activity was increased. Total PDE4 inhibitory activity was slightly decreased in smokers. None of these changes were considered to be clinically meaningful. No dose adjustment is recommended in these patients. A combination of factors, such as in black, non-smoking females, might lead to an increase of exposure and persistent intolerability. In this case, roflumilast treatment should be reassessed (see Precautions).
In study RO-2455-404-RD when compared with the overall population, the total PDE4 inhibitory activity determined from
ex vivo unbound fractions was found to be 15% higher in patients ≥75 years of age, and 11% higher in patients with baseline body weight <60 kg (refer to Precautions).
Renal impairment: Total PDE4 inhibitory activity decreased by 9% in patients with severe renal impairment (creatinine clearance 10-30 ml/min). No dose adjustment is necessary.
Hepatic impairment: The pharmacokinetics of roflumilast 250 micrograms once-daily was tested in 16 patients with mild to moderate hepatic impairment classified as Child-Pugh A and B. In these patients, the total PDE4 inhibitory activity was increased by about 20% in patients with Child-Pugh A and about 90% in patients with Child-Pugh B. Simulations suggest dose proportionality between roflumilast 250 and 500 micrograms in patients with mild and moderate hepatic impairment. Caution is necessary in Child-Pugh A patients (see Dosage & Administration). Patients with moderate or severe hepatic impairment classified as Child-Pugh B or C should not take roflumilast (see Contraindications).
Toxicology: Preclinical safety data: There is no evidence for an immunotoxic, skin sensitising or phototoxic potential.
A slight reduction in male fertility was seen in conjunction with epididymal toxicity in rats. No epididymal toxicity or changes in semen parameters were present in any other rodent or non-rodent species including monkeys in spite of higher exposures.
In one of two rat embryofetal development studies, a higher incidence of incomplete skull bone ossification was seen at a dose producing maternal toxicity. In one of three rat studies on fertility and embryofetal development, post-implantation losses were observed. Post-implantation losses were not seen in rabbits. Prolongation of gestation was seen in mice.
The relevance of these findings to humans is unknown.
Most relevant findings in safety pharmacology and toxicology studies occurred at higher doses and exposure than that intended for clinical use. These findings consisted mainly of gastrointestinal findings (i.e. vomiting, increased gastric secretion, gastric erosions, intestine inflammation) and cardiac findings (i.e. focal haemorrhages, haemosiderin deposits and lympho-histiocytic cell infiltration in the right atria in dogs, and decreased blood pressure and increased heart rate in rats, guinea pigs and dogs).
Rodent-specific toxicity in the nasal mucosa was observed in repeat-dose toxicity and carcinogenicity studies. This effect seems to be due to an ADCP (4-Amino-3,5-dichloro-pyridine) N-oxide intermediate specifically formed in rodent olfactory mucosa, with special binding affinity in these species (i.e. mouse, rat and hamster).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image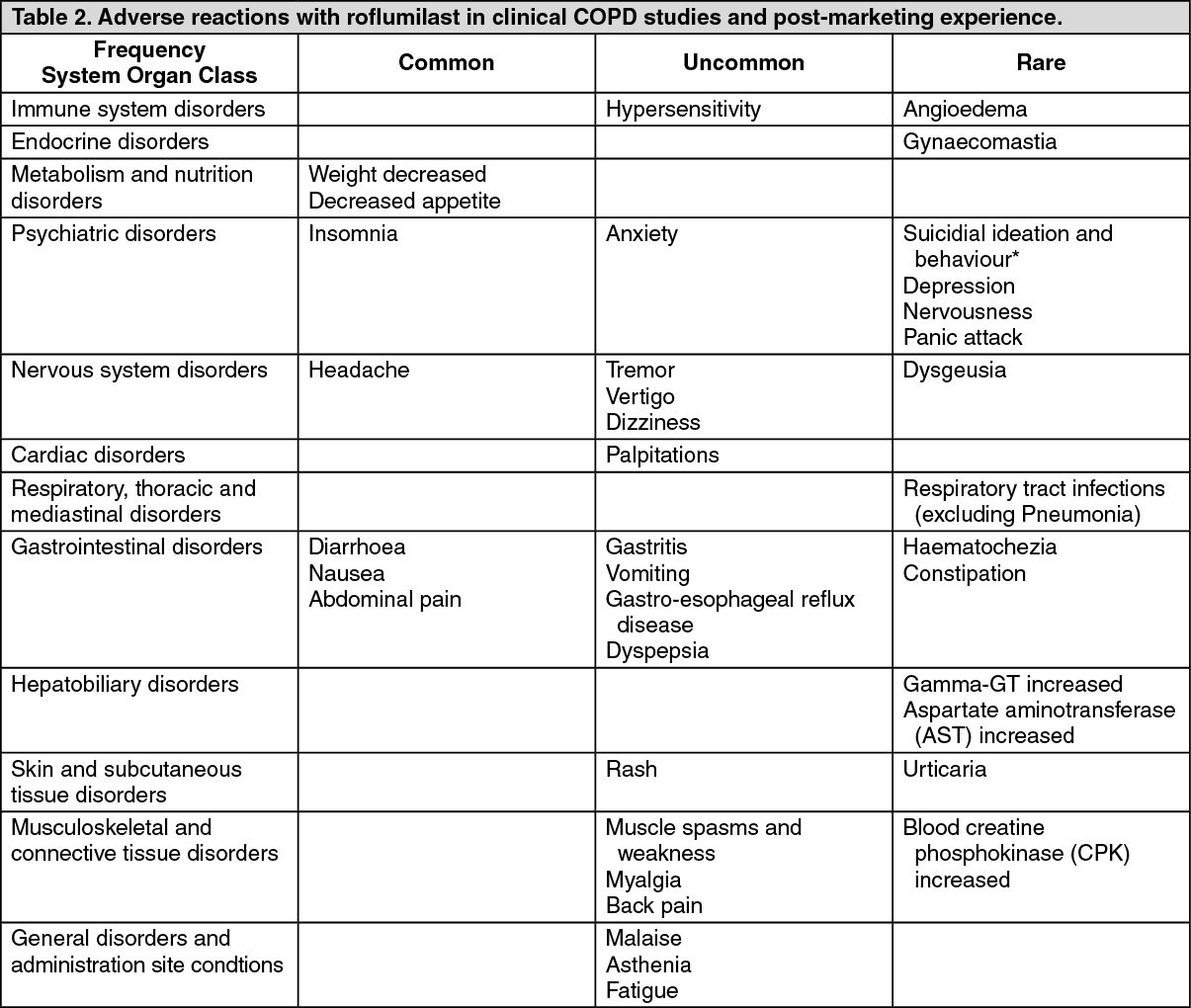 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out