


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Aflibercept is a recombinant fusion protein consisting of portions of human VEGF receptor 1 and 2 extracellular domains fused to the Fc portion of human IgG1.
Aflibercept is produced in Chinese hamster ovary (CHO) K1 cells by recombinant DNA technology.
Aflibercept acts as a soluble decoy receptor that binds VEGF-A and PlGF with higher affinity than their natural receptors, and thereby can inhibit the binding and activation of these cognate VEGF receptors.
Mechanism of action: Vascular endothelial growth factor-A (VEGF-A) and placental growth factor (PlGF) are members of the VEGF family of angiogenic factors that can act as potent mitogenic, chemotactic, and vascular permeability factors for endothelial cells. VEGF acts via two receptor tyrosine kinases; VEGFR-1 and VEGFR-2, present on the surface of endothelial cells. PlGF binds only to VEGFR-1, which is also present on the surface of leucocytes. Excessive activation of these receptors by VEGF-A can result in pathological neovascularisation and excessive vascular permeability. PlGF can synergize with VEGF-A in these processes, and is also known to promote leucocyte infiltration and vascular inflammation.
Pharmacodynamic effects: wet AMD: Wet AMD is characterised by pathological choroidal neovascularisation (CNV). Leakage of blood and fluid from CNV may cause retinal thickening or oedema and/or sub-/intra-retinal haemorrhage, resulting in loss of visual acuity.
In patients treated with Eylea (one injection per month for three consecutive months, followed by one injection every 2 months), central retinal thickness [CRT] decreased soon after treatment initiation, and the mean CNV lesion size was reduced, consistent with the results seen with ranibizumab 0.5 mg every month.
In the VIEW1 study there were mean decreases in CRT on optical coherence tomography (OCT) (-130 and -129 microns at week 52 for the Eylea 2 mg every two months and ranibizumab 0.5 mg every month study groups, respectively). Also at the 52 week time point, in the VIEW2 study there were mean decreases in CRT on OCT (-149 and -139 microns for the Eylea 2 mg every two months and ranibizumab 0.5 mg every month study groups, respectively). The reduction of CNV size and reduction in CRT were generally maintained in the second year of the studies.
The ALTAIR study was conducted in Japanese patients with treatment naïve wet AMD, showing similar outcomes to the VIEW studies using 3 initial monthly Eylea 2 mg injections, followed by one injection after a further 2 months, and then continued with a treat-and-extend regimen with variable treatment intervals (2-week or 4-week adjustments) up to a maximum 16 week interval according to pre-specified criteria. At week 52, there were mean decreases in central retinal thickness (CRT) on OCT of -134.4 and -126.1 microns for the 2-week adjustment group and the 4-week adjustment group, respectively. The proportion of patients without fluid on OCT at week 52 was 68.3% and 69.1% in the 2- and 4-week adjustment groups, respectively. The reduction in CRT was generally maintained in both treatment arms in the second year of the ALTAIR study.
The ARIES study was designed to explore the non-inferiority of an Eylea 2 mg treat-and-extend dosing regimen initiated immediately after administration of 3 initial monthly injections and one additional injection after 2 months vs. a treat-and-extend dosing regimen initiated after one year of treatment. For patients requiring a more frequent than Q8 dosing at least once over the course of the study, CRT remained higher, but the mean decrease in CRT from baseline to week 104 was -160.4 microns, similar to the patients treated at Q8 or less frequent intervals.
Macular oedema secondary to CRVO and BRVO: In CRVO and BRVO, retinal ischaemia occurs and signals the release of VEGF which in turn destabilises the tight junctions, and promotes endothelial cell proliferation. Up-regulation of VEGF is associated with the breakdown of the blood retina barrier, increased vascular permeability, retinal oedema, and neovascularisation complications.
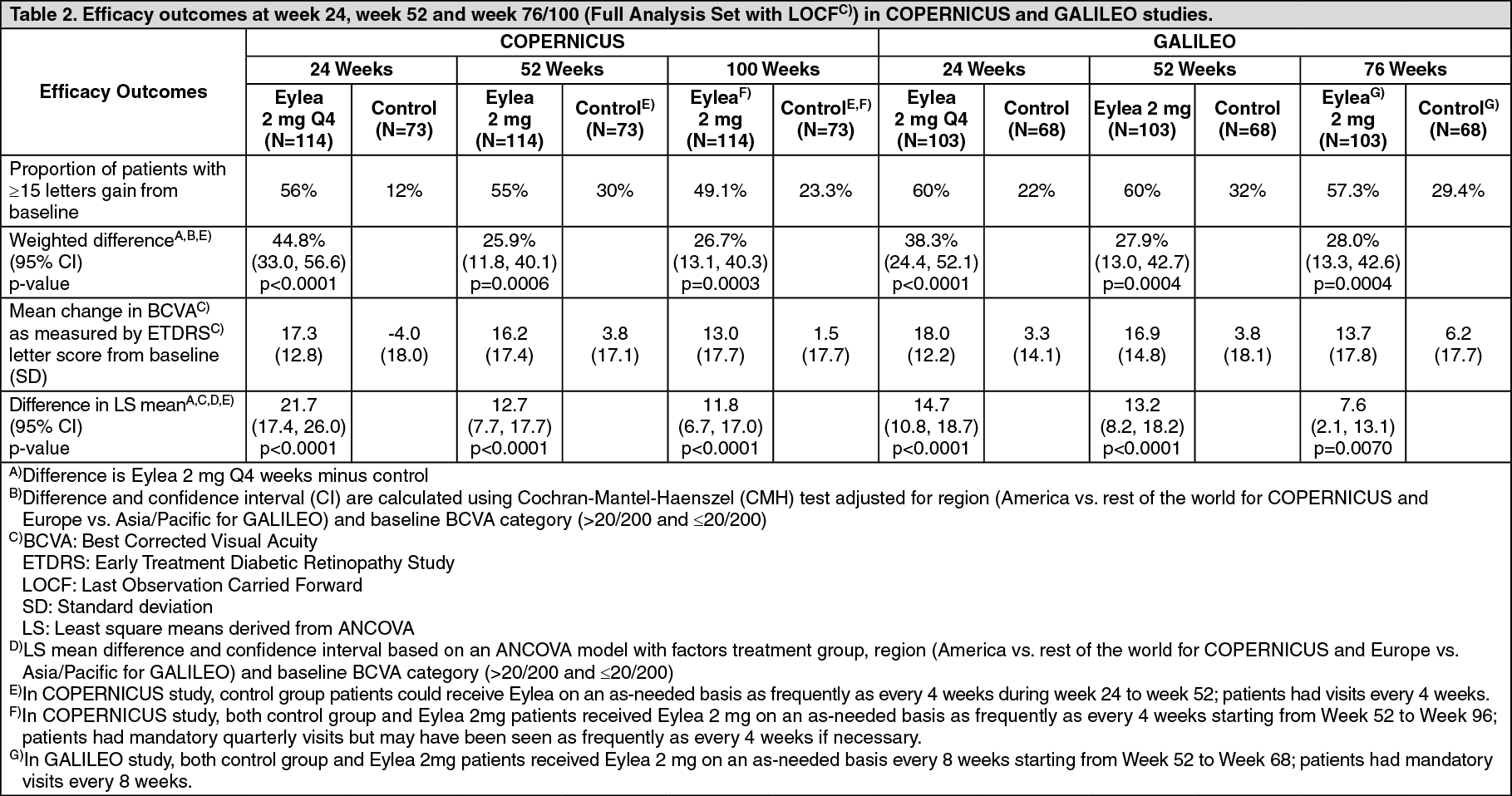
In patients treated with 6 consecutive monthly injections of Eylea 2 mg, there was a consistent, rapid and robust morphologic response (as measured by improvements in mean CRT) observed. At week 24, the reduction in CRT was statistically superior versus control in all three studies (COPERNICUS in CRVO: -457 vs. -145 microns; GALILEO in CRVO: -449 vs. -169 microns; VIBRANT in BRVO: -280 vs. -128 microns). This decrease from baseline in CRT was maintained to the end of each study, week 100 in COPERNICUS, week 76 in GALILEO, and week 52 in VIBRANT.
Diabetic macular oedema: Diabetic macular oedema is a consequence of diabetic retinopathy and is characterised by increased vasopermeability and damage to the retinal capillaries which may result in loss of visual acuity.
In patients treated with Eylea, the majority of whom were classified as having Type II diabetes, a rapid and robust response in morphology (CRT, DRSS level) was observed.
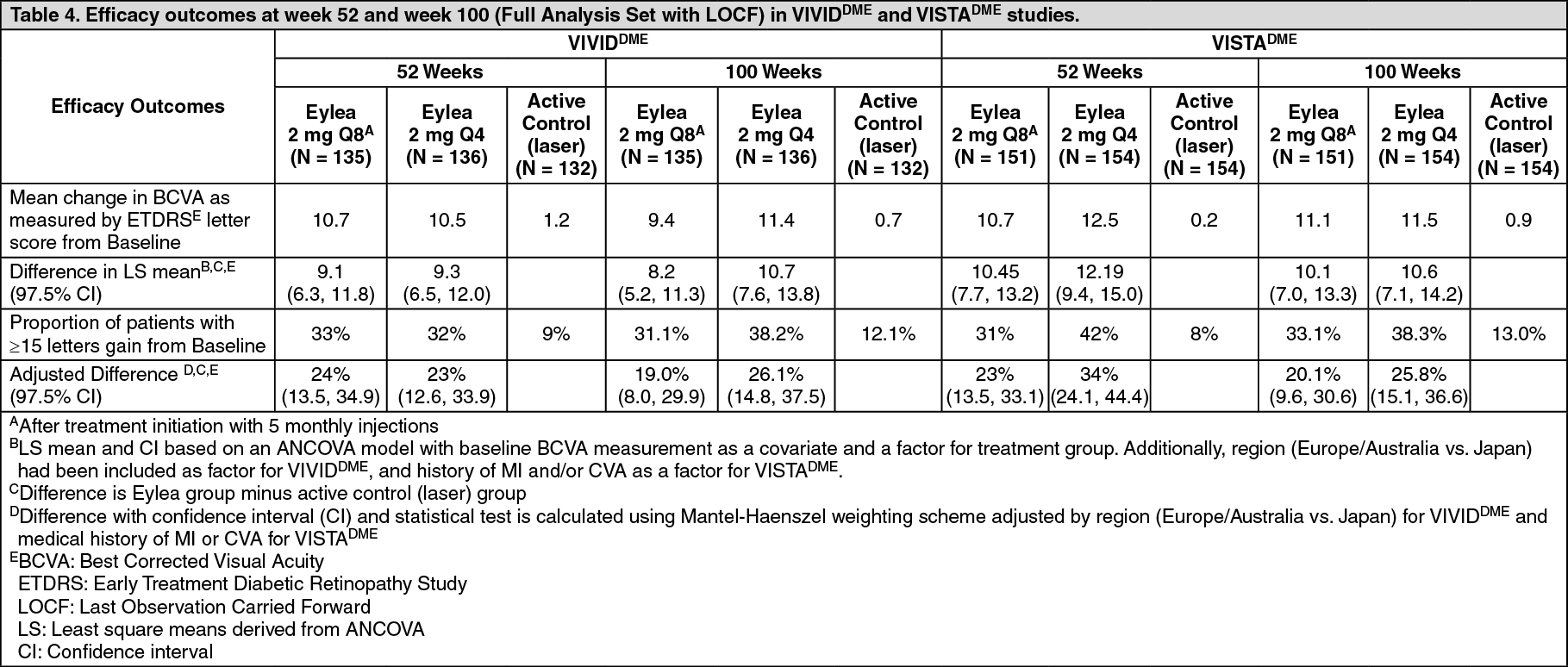
In the VIVIDDME and the VISTADME studies, a statistically significant greater mean decrease in CRT from baseline to week 52 was observed in patients treated with Eylea than with the laser control, -192.4 and -183.1 microns for the 2Q8 Eylea groups and -66.2 and -73.3 microns for the control groups, respectively. At week 100 the decrease was maintained with -195.8 and -191.1 microns for the 2Q8 Eylea groups and -85.7 and -83.9 microns for the control groups, in the VIVIDDME and VISTADME studies, respectively.
A ≥2 step improvement in DRSS was assessed in a pre-specified manner in VIVIDDME and VISTADME. The DRSS score was gradable in 73.7% of the patients in VIVIDDME and 98.3% of the patients in VISTADME. At week 52, 27.7% and 29.1% of the Eylea 2Q8 groups, and 7.5% and 14.3% of the control groups experienced a ≥2 step improvement in the DRSS. At week 100, the respective percentages were 32.6% and 37.1% of the Eylea 2Q8 groups and 8.2% and 15.6% of the control groups.
The VIOLET study compared three different dosing regimens of Eylea 2 mg for treatment of DME after at least one year of treatment at fixed intervals, where treatment was initiated with 5 consecutive monthly doses followed by dosing every 2 months. At week 52 and week 100 of the study, i.e. second and third year of treatment, the mean changes in CRT were clinically similar for treat-and-extend (2T&E), pro re nata (2PRN) and 2Q8, respectively, -2.1, 2.2 and -18.8 microns at week 52, and 2.3, -13.9 and -15.5 microns at week 100.
Myopic choroidal neovascularisation: Myopic choroidal neovascularisation (myopic CNV) is a frequent cause of vision loss in adults with pathologic myopia. It develops as a wound healing mechanism consequent to Bruch's membrane ruptures and represents the most vision-threatening event in pathologic myopia.
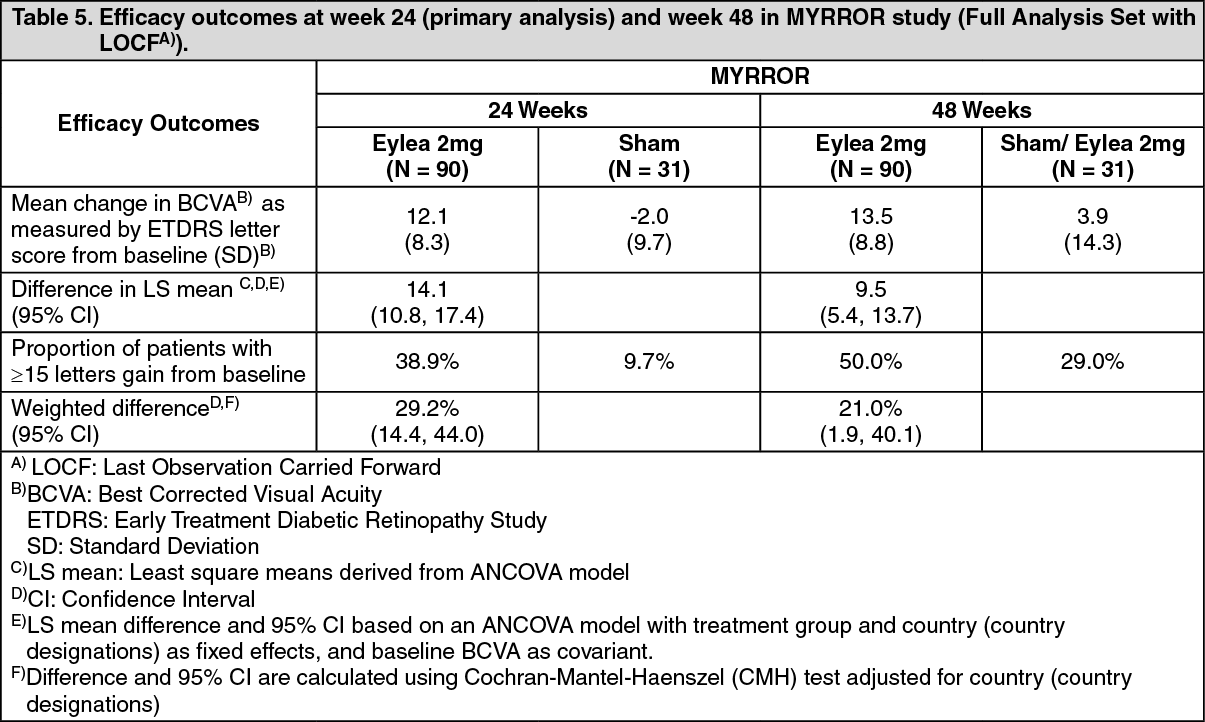
In patients treated with Eylea in the MYRROR study (one injection given at start of therapy, with additional injections given in case of disease persistence or recurrence), CRT decreased soon after treatment initiation favouring Eylea at week 24 (-79 microns and -4 microns for the Eylea 2 mg treatment group and the control group, respectively), which was maintained through week 48. In addition, the mean CNV lesion size decreased.
Clinical efficacy and safety: wet AMD: The safety and efficacy of Eylea were assessed in two randomised, multi-centre, double-masked, active-controlled studies in patients with wet AMD (VIEW1 and VIEW2) with a total of 2,412 patients treated and evaluable for efficacy (1,817 with Eylea). Patient ages ranged from 49 to 99 years with a mean of 76 years. In these clinical studies, approximately 89% (1,616/1,817) of the patients randomised to treatment with Eylea were 65 years of age or older, and approximately 63% (1,139/1,817) were 75 years of age or older. In each study, patients were randomly assigned in a 1:1:1:1 ratio to 1 of 4 dosing regimens: 1) Eylea administered at 2 mg every 8 weeks following 3 initial monthly doses (Eylea 2Q8); 2) Eylea administered at 2 mg every 4 weeks (Eylea 2Q4); 3) Eylea administered at 0.5 mg every 4 weeks (Eylea 0.5Q4); and 4) ranibizumab administered at 0.5 mg every 4 weeks (ranibizumab 0.5Q4).
In the second year of the studies, patients continued to receive the initially randomised dosage but on a modified dosing schedule guided by assessment of visual and anatomic outcomes with a protocol-defined maximum dosing interval of 12 weeks.
In both studies, the primary efficacy endpoint was the proportion of patients in the Per Protocol Set who maintained vision, i.e. losing fewer than 15 letters of visual acuity at week 52 from baseline.
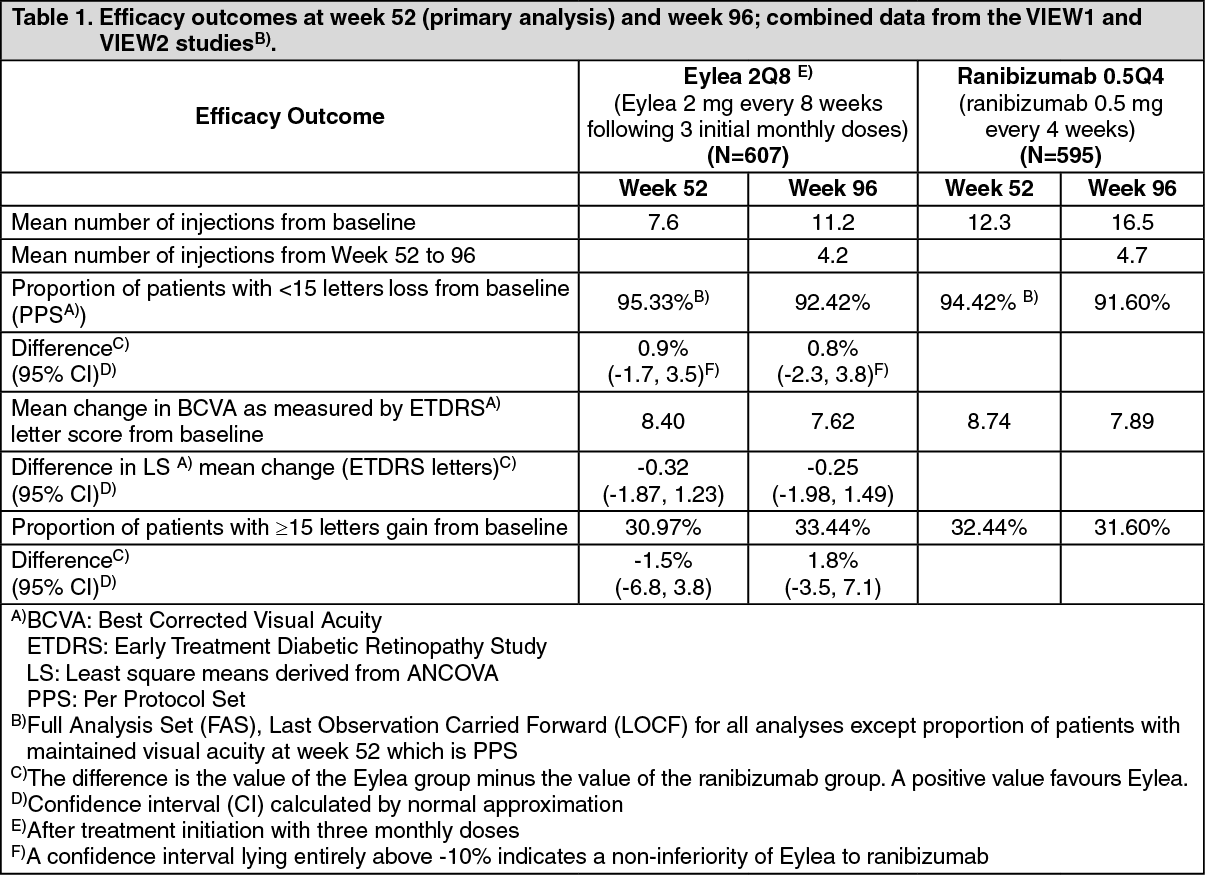
In the VIEW1 study, at week 52, 95.1% of patients in the Eylea 2Q8 group maintained vision compared to 94.4% patients in the ranibizumab 0.5Q4 group. In the VIEW2 study, at week 52, 95.6% of patients in the Eylea 2Q8 group maintained vision compared to 94.4% patients in the ranibizumab 0.5Q4 group. In both studies Eylea was shown to be non-inferior and clinically equivalent to the ranibizumab 0.5Q4 group.
Detailed results from the combined analysis of both studies are shown in Table 1 and Figure 1 as follows. (See Table 1 and Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image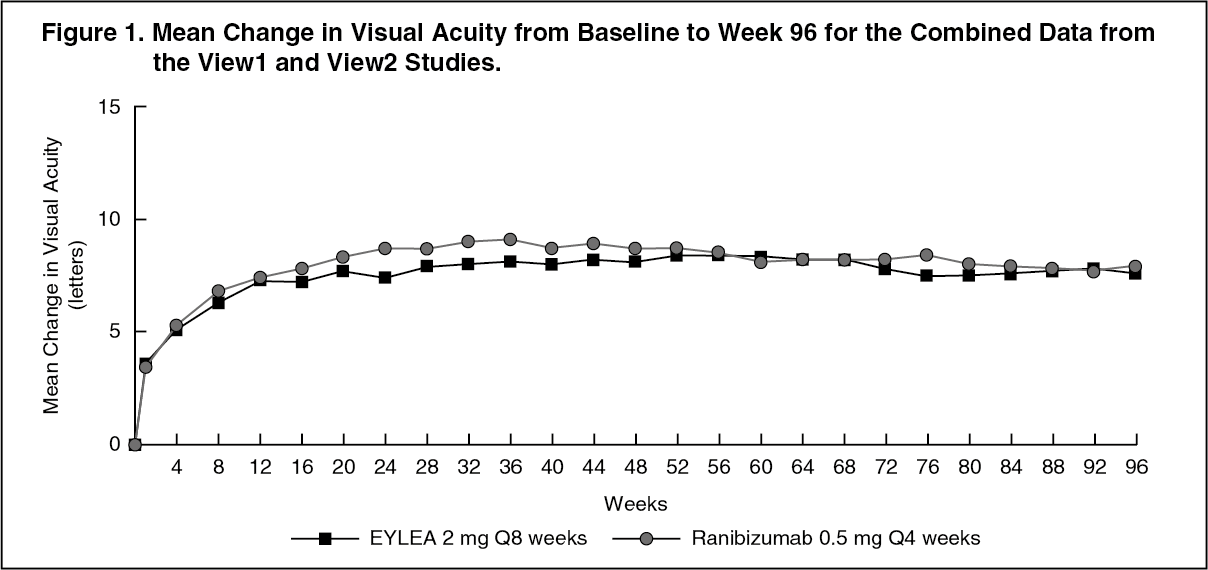 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn combined data analysis of VIEW1 and VIEW2, Eylea demonstrated clinically meaningful changes from baseline in pre-specified secondary efficacy endpoint National Eye Institute Visual Function Questionnaire (NEI VFQ-25) without clinically meaningful differences to ranibizumab. The magnitude of these changes was similar to that seen in published studies, which corresponded to a 15-letter gain in Best Corrected Visual Acuity (BCVA).
In the second year of the studies, efficacy was generally maintained through the last assessment at week 96, and 2-4% of patients required all injections on a monthly basis, and a third of patients required at least one injection with a treatment interval of only one month.
Decreases in mean CNV area were evident in all dose groups in both studies.
Efficacy results in all evaluable subgroups (e.g. age, gender, race, baseline visual acuity, lesion type, lesion size) in each study and in the combined analysis were consistent with the results in the overall populations.
ALTAIR was a 96 week multicentre, randomised, open-label study in 247 Japanese patients with treatment naïve wet AMD, designed to assess the efficacy and safety of Eylea following two different adjustment intervals (2-weeks and 4-weeks) of a treat-and-extend dosing regimen.
All patients received monthly doses of Eylea 2 mg for 3 months, followed by one injection after a further 2 month interval. At week 16, patients were randomised 1:1 into two treatment groups: 1) Eylea treat-and-extend with 2-week adjustments and 2) Eylea treat-and-extend with 4-week adjustments. Extension or shortening of the treatment interval was decided based on visual and/or anatomic criteria defined by protocol with a maximum treatment interval of 16 weeks for both groups.
The primary efficacy endpoint was mean change in BCVA from baseline to week 52. The secondary efficacy endpoints were the proportion of patients who did not lose ≥15 letters and the proportion of patients who gained at least 15 letters of BCVA from baseline to week 52.
At week 52, patients in the treat-and-extend arm with 2-week adjustments gained a mean of 9.0 letters from baseline as compared to 8.4 letters for those in the 4-week adjustment group [LS mean difference in letters (95% CI): -0.4 (-3.8, 3.0), ANCOVA]. The proportion of patients who did not lose ≥15 letters in the two treatment arms was similar (96.7% in the 2-week and 95.9% in the 4-week adjustment groups). The proportion of patients who gained ≥15 letters at week 52 was 32.5% in the 2-week adjustment group and 30.9% in the 4-week adjustment group. The proportion of patients who extended their treatment interval to 12 weeks or beyond was 42.3% in the 2-week adjustment group and 49.6% in the 4-week adjustment group. Furthermore, in the 4-week adjustment group 40.7% of patients were extended to 16 week intervals. At the last visit up to week 52, 56.8% and 57.8% of patients in the 2-week and 4-week adjustment groups, respectively had their next injection scheduled at an interval of 12 weeks or beyond.
In the second year of the study, efficacy was generally maintained up to and including the last assessment at week 96, with a mean gain from baseline of 7.6 letters for the 2-week adjustment group and 6.1 letters for the 4-week adjustment group. The proportion of patients who extended their treatment interval to 12 weeks or beyond was 56.9% in the 2-week adjustment group and 60.2% in the 4-week adjustment group. At the last visit prior to week 96, 64.9% and 61.2% of patients in the 2-week and 4-week adjustment groups, respectively had their next injection scheduled at an interval of 12 weeks or beyond. During the second year of treatment patients in both the 2-week and 4-week adjustment groups received an average of 3.6 and 3.7 injections, respectively. Over the 2 year treatment period patients received an average of 10.4 injections.
Ocular and systemic safety profiles were similar to the safety observed in the pivotal studies VIEW1 and VIEW2.
ARIES was a 104-week multicentre, randomised, open-label, active-controlled study in 269 patients with treatment naïve wet AMD, designed to assess the non-inferiority in terms of efficacy as well as the safety of a treat-and-extend dosing regimen initiated after 3 consecutive monthly doses followed by extension to a 2 monthly treatment interval vs. a treat-and-extend dosing regimen initiated after the first year of treatment.
The ARIES study also explored the percentage of patients that required more frequent treatment than every 8 weeks based on the investigator's decision. Out of the 269 patients 62 patients received more frequent dosing at least once during the course of the study. Such patients remained in the study and received treatment according to the investigator's best clinical judgement but not more frequently than every 4 weeks and their treatment intervals could be extended again afterwards. The average treatment interval after the decision to treat more frequently was 6.1 weeks. Week 104 BCVA was lower in patients requiring more intensive treatment at least once over the course of the study compared with patients who did not and the mean change in BCVA from baseline to end of the study was +2.3 ± 15.6 letters. Among the patients treated more frequently, 85.5% maintained vision, i.e. lost less than 15 letters, and 19.4% gained 15 letters or more. The safety profile of patients treated more frequently than every 8 weeks was comparable to the safety data in VIEW1 and VIEW2.
Macular oedema secondary to CRVO: The safety and efficacy of Eylea were assessed in two randomised, multi-centre, double-masked, sham-controlled studies in patients with macular oedema secondary to CRVO (COPERNICUS and GALILEO) with a total of 358 patients treated and evaluable for efficacy (217 with Eylea). Patient ages ranged from 22 to 89 years with a mean of 64 years. In the CRVO studies, approximately 52% (112/217) of the patients randomised to treatment with Eylea were 65 years of age or older, and approximately 18% (38/217) were 75 years of age or older. In both studies, patients were randomly assigned in a 3:2 ratio to either 2 mg Eylea administered every 4 weeks (2Q4), or the control group receiving sham injections every 4 weeks for a total of 6 injections.
After 6 consecutive monthly injections, patients received treatment only if they met pre-specified retreatment criteria, except for patients in the control group in the GALILEO study who continued to receive sham (control to control) until week 52. From this timepoint all patients were treated if pre-specified criteria were met.
In both studies, the primary efficacy endpoint was the proportion of patients who gained at least 15 letters in BCVA at week 24 compared to baseline. A secondary efficacy variable was change in visual acuity at week 24 compared to baseline.
The difference between treatment groups was statistically significant in favour of Eylea in both studies. The maximal improvement in visual acuity was achieved at month 3 with subsequent stabilisation of visual acuity and CRT until month 6. The statistically significant difference was maintained through week 52.
Detailed results from the analysis of both studies are shown in Table 2 and Figure 2 as follows. (See Table 2 and Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image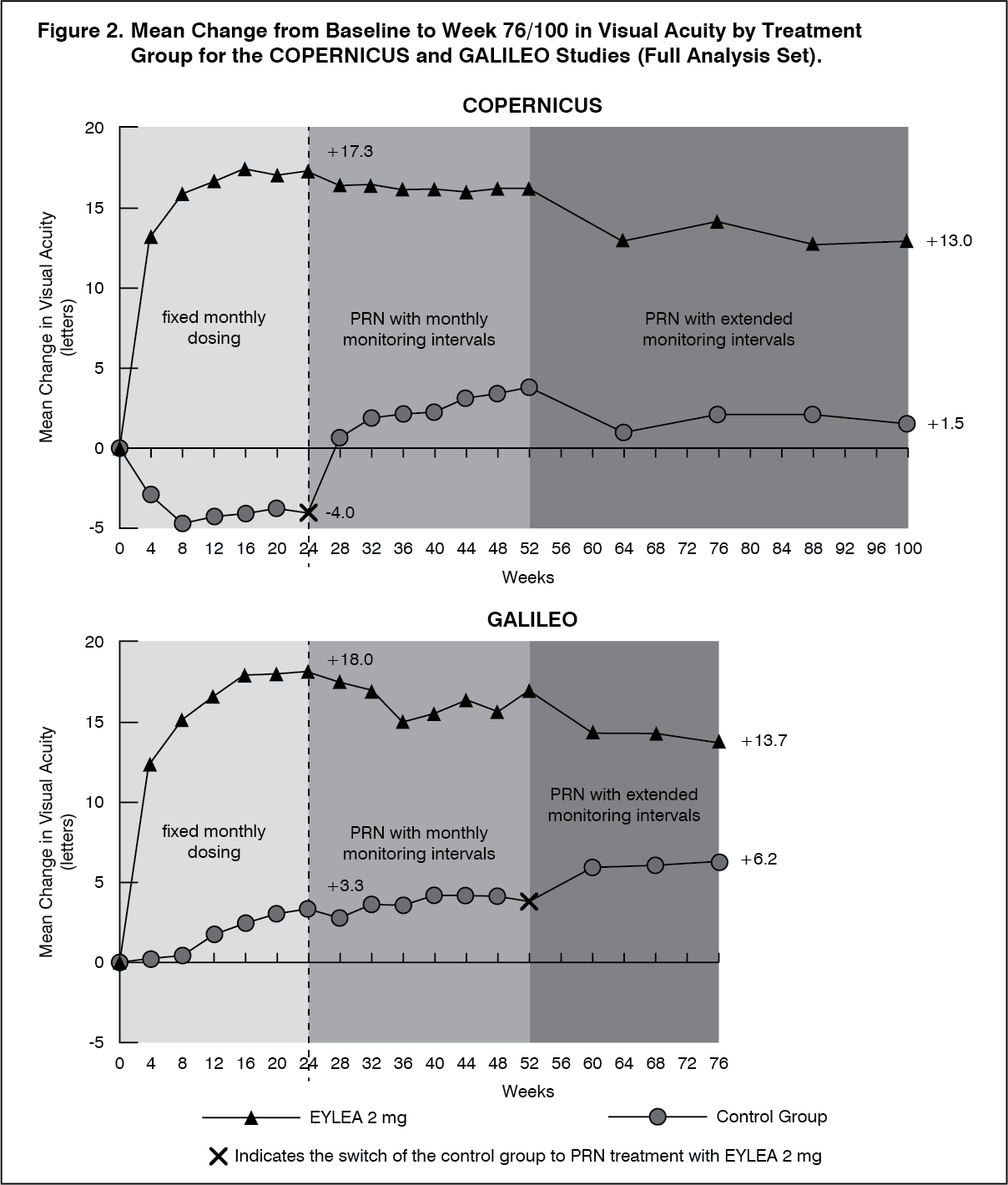 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn GALILEO, 86.4% (n=89) of the Eylea group and 79.4% (n=54) of the sham group had perfused CRVO at baseline. At week 24, this was 91.8% (n=89) in the Eylea group and 85.5% (n=47) in the sham group. These proportions were maintained at week 76, with 84.3% (n=75) in the Eylea group and 84.0% (n=42) in the sham group.
In COPERNICUS, 67.5% (n=77) of the Eylea group and 68.5% (n=50) of the sham group had perfused CRVO at baseline. At week 24, this was 87.4% (n=90) in the Eylea group and 58.6% (n=34) in the sham group. These proportions were maintained at week 100 with 76.8% (n=76) in the Eylea group and 78% (n=39) in the sham group. Patients in the sham group were eligible to receive Eylea from week 24.
The beneficial effect of Eylea treatment on visual function was similar in the baseline subgroups of perfused and non-perfused patients. Treatment effects in other evaluable subgroups (e.g. age, gender, race, baseline visual acuity, CRVO duration) in each study were in general consistent with the results in the overall populations.
In combined data analysis of GALILEO and COPERNICUS, Eylea demonstrated clinically meaningful changes from baseline in pre-specified secondary efficacy endpoint National Eye Institute Visual Function Questionnaire (NEI VFQ-25). The magnitude of these changes was similar to that seen in published studies, which corresponded to a 15-letter gain in Best Corrected Visual Acuity (BCVA).
Macular oedema secondary to BRVO: The safety and efficacy of Eylea were assessed in a randomised, multi-centre, double-masked, active-controlled study in patients with macular oedema secondary to BRVO (VIBRANT) which included Hemi-Retinal Vein Occlusion. A total of 181 patients were treated and evaluable for efficacy (91 with Eylea). Patient ages ranged from 42 to 94 years with a mean of 65 years. In the BRVO study, approximately 58% (53/91) of the patients randomised to treatment with Eylea were 65 years of age or older, and approximately 23% (21/91) were 75 years of age or older. In the study, patients were randomly assigned in a 1:1 ratio to either 2 mg Eylea administered every 8 weeks following 6 initial monthly injections or laser photocoagulation administered at baseline (laser control group). Patients in the laser control group could receive additional laser photocoagulation (called 'rescue laser treatment') beginning at week 12 with a minimum interval of 12 weeks. Based on pre-specified criteria, patients in the laser group could receive rescue treatment with Eylea 2 mg from week 24, administered every 4 weeks for 3 months followed by every 8 weeks.
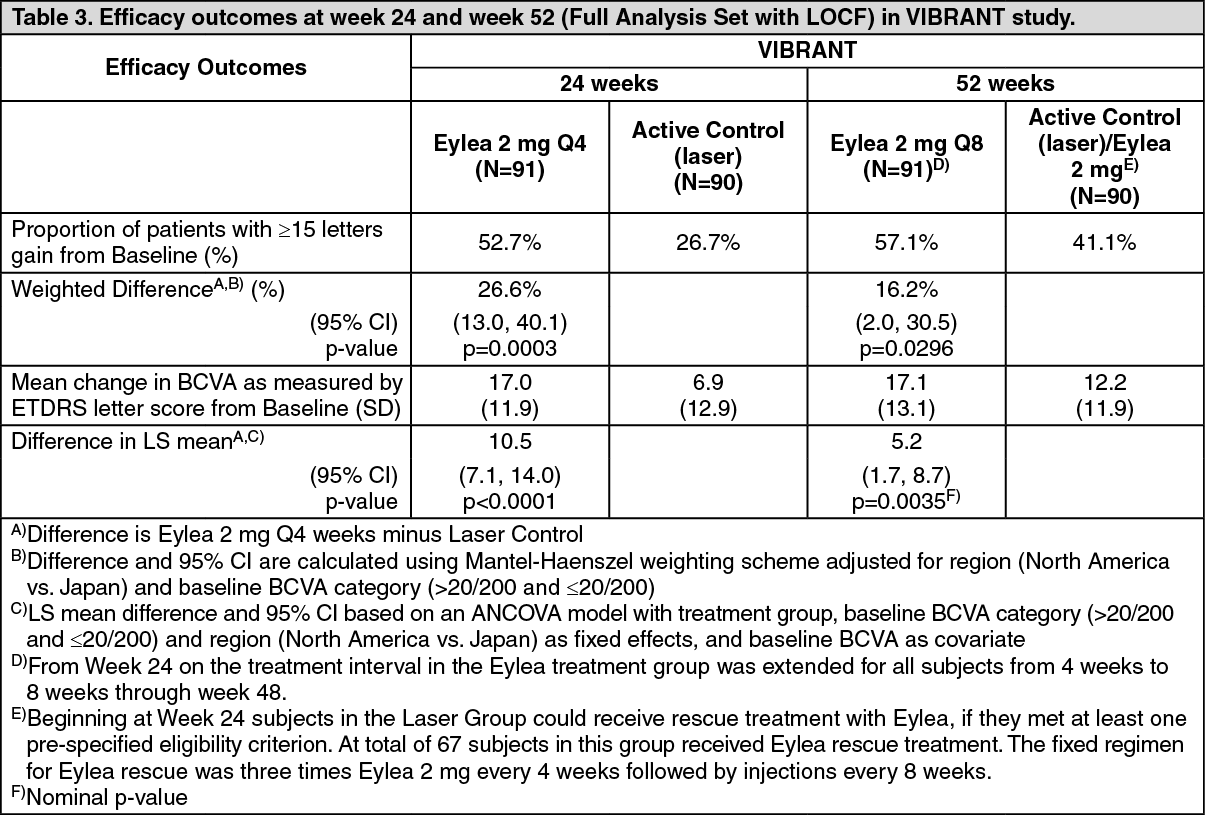
In the VIBRANT study, the primary efficacy endpoint was the proportion of patients who gained at least 15 letters in BCVA at week 24 compared to baseline and the Eylea group was superior to laser control.
A secondary efficacy endpoint was change in visual acuity at week 24 compared to baseline, which was statistically significant in favour of Eylea in the VIBRANT study. The course of visual improvement was rapid and peaked at 3 months with maintenance of the effect until month 12.
In the laser group 67 patients received rescue treatment with Eylea beginning at week 24 (Active Control / Eylea 2 mg group), which resulted in improvement of visual acuity by about 5 letters from week 24 to 52.
Detailed results from the analysis of the VIBRANT study are shown in Table 3 and Figure 3 as follows. (See Table 3 and Figure 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image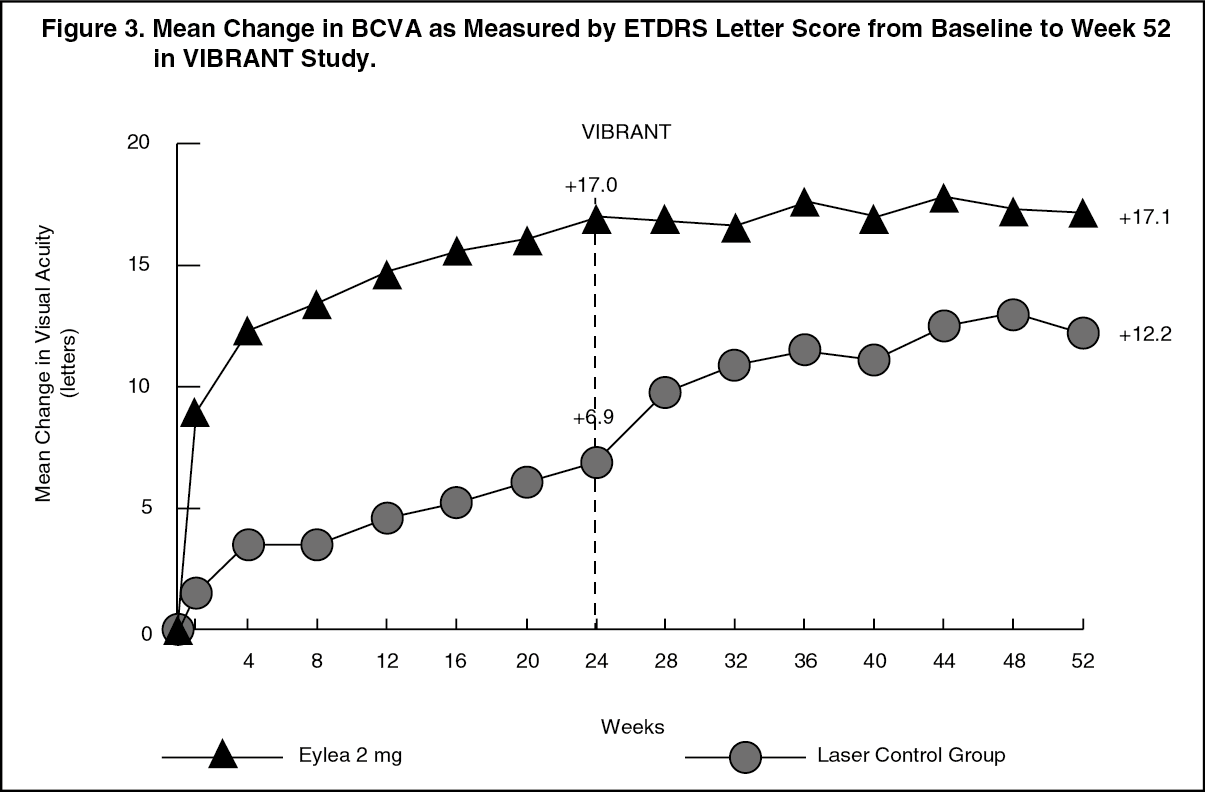 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAt baseline, the proportion of perfused patients in the Eylea and laser groups was 60% and 68%, respectively. At week 24 these proportions were 80% and 67%, respectively. In the Eylea group the proportion of perfused patients was maintained through week 52. In the laser group, where patients were eligible for rescue treatment with Eylea from week 24, the proportion of perfused patients increased to 78% by week 52.
Diabetic macular oedema: The safety and efficacy of Eylea were assessed in two randomised, multi-centre, double-masked, active-controlled studies in patients with DME (VIVIDDME and VISTADME). A total of 862 patients were treated and evaluable for efficacy, 576 with Eylea. Patient ages ranged from 23 to 87 years with a mean of 63 years. In the DME studies, approximately 47% (268/576) of the patients randomised to treatment with Eylea were 65 years of age or older, and approximately 9% (52/576) were 75 years of age or older. The majority of patients in both studies had Type II diabetes.
In both studies, patients were randomly assigned in a 1:1:1 ratio to 1 of 3 dosing regimens: 1) Eylea administered 2 mg every 8 weeks following 5 initial monthly injections (Eylea 2Q8); 2) Eylea administered 2 mg every 4 weeks (Eylea 2Q4); and 3) Macular laser photocoagulation (active control).
Beginning at week 24, patients meeting a pre-specified threshold of vision loss were eligible to receive additional treatment: patients in the Eylea groups could receive laser and patients in the control group could receive Eylea.
In both studies, the primary efficacy endpoint was the mean change from baseline in BCVA at week 52 and both Eylea 2Q8 and Eylea 2Q4 groups demonstrated statistical significance and were superior to the control group. This benefit was maintained through week 100.
Detailed results from the analysis of the VIVIDDME and VISTADME studies are shown in Table 4 and Figure 4 as follows. (See Table 4 and Figure 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image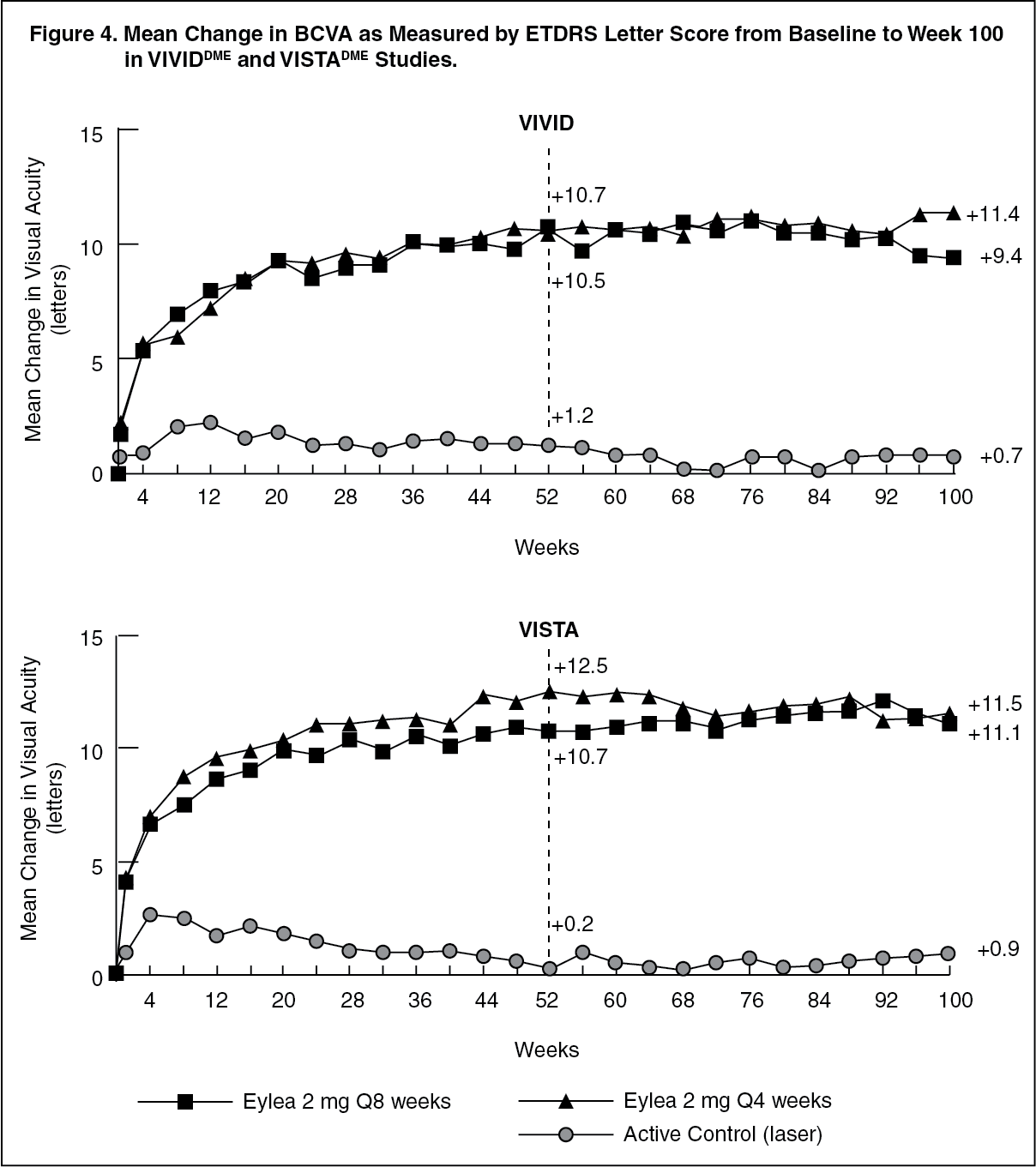 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment effects in evaluable subgroups (e.g., age, gender, race, baseline HbA1c, baseline visual acuity, prior anti-VEGF therapy) in each study and in the combined analysis were generally consistent with the results in the overall populations.
In the VIVIDDME and VISTADME studies, 36 (9%) and 197 (43%) patients received prior anti-VEGF therapy, respectively, with a 3-month or longer washout period. Treatment effects in the subgroup of patients who had previously been treated with a VEGF inhibitor were similar to those seen in patients who were VEGF inhibitor naïve.
Patients with bilateral disease were eligible to receive anti-VEGF treatment in their fellow eye if assessed necessary by the physician. In the VISTADME study, 217 (70.7%) of Eylea patients received bilateral Eylea injections until week 100; in the VIVIDDME study, 97 (35.8%) of Eylea patients received a different anti-VEGF treatment in their fellow eye.
An independent comparative trial (DRCR.net Protocol T) utilised a flexible dosing regimen based on strict OCT and vision re-treatment criteria. In the aflibercept treatment group (n=224) at week 52, this treatment regimen resulted in patients receiving a mean of 9.2 injections, which is similar to the administered number of doses in the Eylea 2Q8 group in VIVIDDME and VISTADME, while overall efficacy of the aflibercept treatment group in Protocol T was comparable to the Eylea 2Q8 group in VIVIDDME and VISTADME. A 13.3 mean letter gain with 42% of patients gaining at least 15 letters in vision from baseline was observed in Protocol T. Safety outcomes demonstrated that overall incidences of ocular and non-ocular adverse events (including ATEs) were comparable across all treatment groups in each of the studies and between the studies.
VIOLET, a 100-week multicentre, randomised, open-label, active controlled study in patients with DME compared three different dosing regimens of Eylea 2 mg for treatment of DME after at least one year of treatment at fixed intervals, where treatment was initiated with 5 consecutive monthly doses followed by dosing every 2 months. The study evaluated non-inferiority of Eylea 2 mg dosed according to a treat-and-extend regimen (2T&E where injections intervals were kept at a minimum of 8 weeks and gradually extended based on clinical and anatomical outcomes) and Eylea 2 mg dosed as needed (2PRN where patients were observed every 4 weeks and injected when needed based on clinical and anatomical outcomes), compared to Eylea 2 mg dosed every 8 weeks (2Q8) for the second and third year of treatment.
The primary efficacy endpoint (change in BCVA from baseline to week 52) was 0.5 ± 6.7 letters in the 2T&E group and 1.7 ± 6.8 letters in the 2PRN group compared to 0.4 ± 6.7 letters in the 2Q8 group, achieving statistical non-inferiority (p<0.0001 for both comparisons; NI margin 4 letters). The changes in BCVA from baseline to week 100 were consistent with the week 52 results: -0.1 ± 9.1 letters in the 2T&E group and 1.8 ± 9.0 letters in the 2PRN group compared to 0.1 ± 7.2 letters in the 2Q8 group. The mean number of injections over 100 weeks were 12.3, 10.0 and 11.5 for 2Q8fix, 2T&E and 2PRN, respectively.
Ocular and systemic safety profiles in all 3 treatment groups were similar to those observed in the pivotal studies VIVID and VISTA.
In the 2T&E group, the increments and decrements for the injection intervals were at the investigator's discretion; increments of 2 weeks were recommended in the study.
Myopic choroidal neovascularisation: The safety and efficacy of Eylea were assessed in a randomised, multi-centre, double-masked, sham-controlled study in treatment-naïve, Asian patients with myopic CNV. A total of 121 patients were treated and evaluable for efficacy (90 with Eylea). Patient ages ranged from 27 to 83 years with a mean of 58 years. In the myopic CNV study, approximately 36% (33/91) of the patients randomised to treatment with Eylea were 65 years of age or older, and approximately 10% (9/91) were 75 years of age or older.
Patients were randomly assigned in a 3:1 ratio to receive either 2 mg Eylea intravitreally or sham injections administered once at study start with additional injections given monthly in case of disease persistence or recurrence until week 24, when the primary endpoint was assessed. At week 24, patients initially randomised to sham were eligible to receive the first dose of Eylea. Following this, patients in both groups continued to be eligible for additional injections in case of disease persistence or recurrence.
The difference between treatment groups was statistically significant in favour of Eylea for the primary endpoint (change in BCVA) and confirmatory secondary efficacy endpoint (proportion of patients who gained 15 letters in BCVA) at week 24 compared to baseline. Differences for both endpoints were maintained through week 48.
Detailed results from the analysis of the MYRROR study are shown in Table 5 and Figure 5 as follows. (See Table 5 and Figure 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image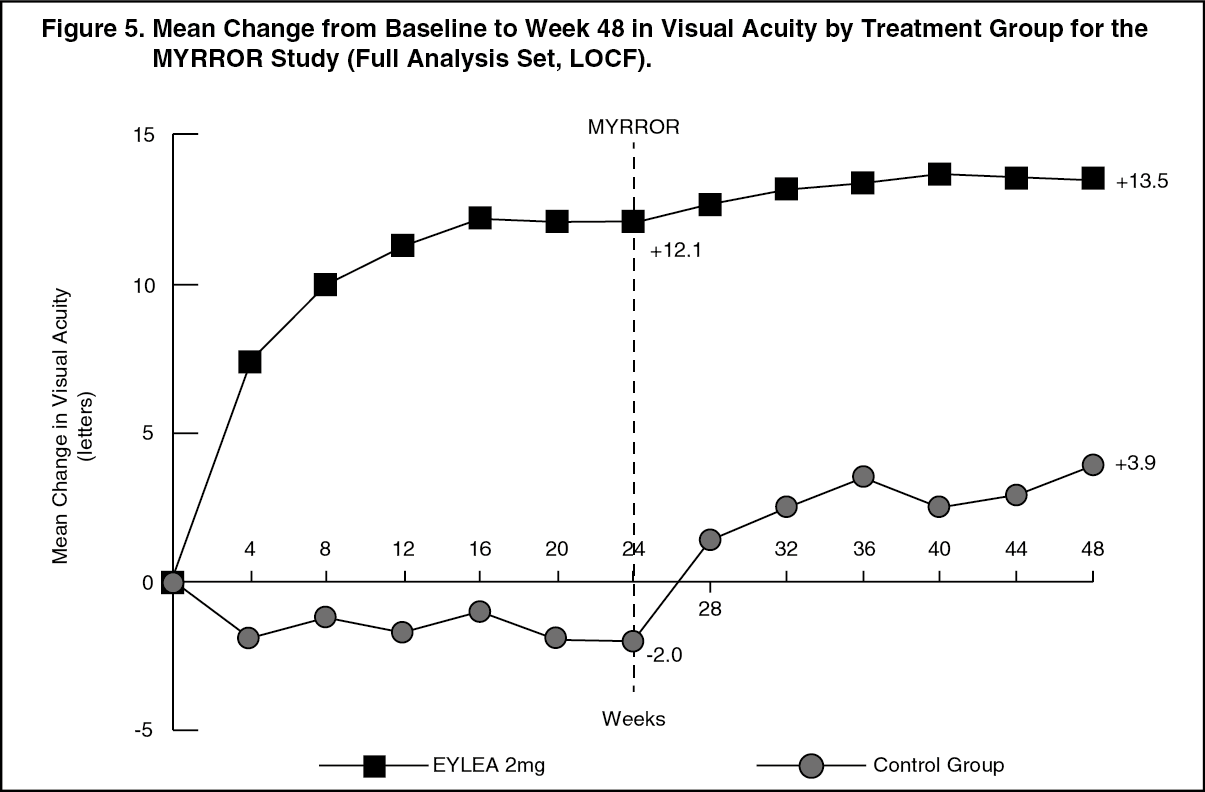 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePaediatric population: The European Medicines Agency has waived the obligation to submit the results of studies with Eylea in all subsets of the paediatric population in wet AMD, CRVO, BRVO, DME and myopic CNV populations (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: Eylea is administered directly into the vitreous to exert local effects in the eye.
Absorption / Distribution: Aflibercept is slowly absorbed from the eye into the systemic circulation after intravitreal administration and is predominately observed in the systemic circulation as an inactive, stable complex with VEGF; however only "free aflibercept" is able to bind endogenous VEGF.
In a pharmacokinetic sub-study in 6 neovascular wet AMD patients with frequent sampling, maximum plasma concentrations of free aflibercept (systemic Cmax) were low, with a mean of approximately 0.02 microgram/mL (range 0 to 0.054) within 1 to 3 days after a 2 mg intravitreal injection, and were undetectable two weeks following dosage in almost all patients. Aflibercept does not accumulate in the plasma when administered intravitreally every 4 weeks.
The mean maximum plasma concentration of free aflibercept is approximately 50 to 500 times below the aflibercept concentration required to inhibit the biologic activity of systemic VEGF by 50% in animal models, in which blood pressure changes were observed after circulating levels of free aflibercept attained approximately 10 microgram/mL and returned to baseline when levels fell below approximately 1 microgram/mL. It is estimated that after intravitreal administration of 2 mg to patients, the mean maximum plasma concentration of free aflibercept is more than 100-fold lower than the concentration of aflibercept required to half-maximally bind systemic VEGF (2.91 microgram/mL) in a study of healthy volunteers. Therefore, systemic pharmacodynamic effects such as blood pressure changes are unlikely.
In pharmacokinetic sub-studies in patients with CRVO, BRVO, DME or myopic CNV mean Cmax of free aflibercept in plasma were similar with values in the range of 0.03 to 0.05 microgram/mL and individual values not exceeding 0.14 microgram/mL. Thereafter, plasma concentrations of free aflibercept declined to values below or close to the lower limit of quantitation generally within one week; undetectable concentrations were reached before the next administration after 4 weeks in all patients.
Elimination: As Eylea is a protein-based therapeutic, no metabolism studies have been conducted.
Free aflibercept binds VEGF to form a stable, inert complex. As with other large proteins, both free and bound aflibercept are expected to be cleared by proteolytic catabolism.
Renal impairment: No special studies in patients with renal impairment have been conducted with Eylea.
Pharmacokinetic analysis of patients in the VIEW2 study, of which 40% had renal impairment (24% mild, 15% moderate, and 1% severe), revealed no differences with respect to plasma concentrations of active drug after intravitreal administration every 4 or 8 weeks.
Similar results were seen in patients with CRVO in the GALILEO study, in patients with DME in the VIVIDDME study, and in patients with myopic CNV in the MYRROR study.
Toxicology: Preclinical safety data: Effects in non-clinical studies on repeated dose toxicity were observed only at systemic exposures considered substantially in excess of the maximum human exposure after intravitreal administration at the intended clinical dose indicating little relevance to clinical use.
Erosions and ulcerations of the respiratory epithelium in nasal turbinates in monkeys treated with aflibercept intravitreally were observed at systemic exposures in excess of the maximum human exposure. The systemic exposure based on Cmax and AUC for free aflibercept were approximately 200- and 700-fold higher, respectively, when compared to corresponding values observed in humans after an intravitreal dose of 2 mg. At the No Observed Adverse Effect Level (NOAEL) of 0.5 mg/eye in monkeys the systemic exposure was 42- and 56-fold higher based on Cmax and AUC, respectively.
No studies have been conducted on the mutagenic or carcinogenic potential of aflibercept.
An effect of aflibercept on intrauterine development was shown in embryo-foetal development studies in pregnant rabbits with intravenous (3 to 60 mg/kg) as well as subcutaneous (0.1 to 1 mg/kg) administration. The maternal NOAEL was at the dose of 3 mg/kg or 1 mg/kg, respectively. A developmental NOAEL was not identified. At the 0.1 mg/kg dose, the systemic exposures based on Cmax and cumulative AUC for free aflibercept were approximately 17- and 10-fold higher, respectively, when compared to corresponding values observed in humans after an intravitreal dose of 2 mg.
Effects on male and female fertility were assessed as part of a 6-month study in monkeys with intravenous administration of aflibercept at doses ranging from 3 to 30 mg/kg. Absent or irregular menses associated with alterations in female reproductive hormone levels and changes in sperm morphology and motility were observed at all dose levels. Based on Cmax and AUC for free aflibercept observed at the 3 mg/kg intravenous dose, the systemic exposures were approximately 4,900-fold and 1,500-fold higher, respectively, than the exposure observed in humans after an intravitreal dose of 2 mg. All changes were reversible.