Pharmacotherapeutic group: Antiviral for systemic use; antivirals for treatment of HIV infections, combinations.
ATC code: J05AR19.
Pharmacology: Pharmacodynamics: Mechanism of action and pharmacodynamic effects: Emtricitabine is a nucleoside reverse transcriptase inhibitor (NRTI) and analogue of 2'-deoxycytidine. Emtricitabine is phosphorylated by cellular enzymes to form emtricitabine triphosphate. Emtricitabine triphosphate competitively inhibits HIV-1 reverse transcriptase (RT), resulting in deoxyribonucleic acid (DNA) chain termination. Emtricitabine has activity against HIV-1, HIV-2, and HBV.
Rilpivirine is a diarylpyrimidine NNRTI of HIV-1. Rilpivirine activity is mediated by non-competitive inhibition of HIV-1 RT. Rilpivirine does not inhibit the human cellular DNA polymerases α, β and mitochondrial DNA polymerase γ.
Tenofovir alafenamide is a nucleotide reverse transcriptase inhibitor (NtRTI) and prodrug of tenofovir (2'-deoxyadenosine monophosphate analogue). Due to increased plasma stability and intracellular activation through hydrolysis by cathepsin A, tenofovir alafenamide is more efficient than tenofovir disoproxil fumarate in loading tenofovir into peripheral blood mononuclear cells (PBMCs) (including lymphocytes and other HIV target cells) and macrophages. Intracellular tenofovir is subsequently phosphorylated to the active metabolite tenofovir diphosphate. Tenofovir diphosphate inhibits HIV RT, resulting in DNA chain termination. Tenofovir has activity against HIV-1, HIV-2 and HBV.
Antiviral activity in vitro: The combinations of emtricitabine, rilpivirine, and tenofovir alafenamide were not antagonistic and showed synergistic effects with each other in cell culture combination antiviral activity assays.
The antiviral activity of emtricitabine against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, the MAGI CCR5 cell line, and PBMCs. The 50% effective concentration (EC
50) values for emtricitabine were in the range of 0.0013 to 0.64 μM. Emtricitabine displayed antiviral activity in cell culture against HIV-1 subtype A, B, C, D, E, F, and G (EC
50 values ranged from 0.007 to 0.075 μM) and showed activity against HIV-2 (EC
50 values ranged from 0.007 to 1.5 μM).
Rilpivirine exhibited activity against laboratory strains of wild-type HIV-1 in an acutely infected T-cell line with a median EC
50 value for HIV-1/IIIB of 0.73 nM (0.27 ng/mL). Rilpivirine also demonstrated antiviral activity against a broad panel of HIV-1 group M (subtype A, B, C, D, F, G, H) primary isolates with EC
50 values ranging from 0.07 to 1.01 nM (0.03 to 0.37 ng/mL), group O primary isolates with EC
50 values ranging from 2.88 to 8.45 nM (1.06 to 3.10 ng/mL), and showed limited
in vitro activity against HIV-2 with EC
50 values ranging from 2,510 to 10,830 nM (920 to 3,970 ng/mL).
The antiviral activity of tenofovir alafenamide against laboratory and clinical isolates of HIV-1 subtype B was assessed in lymphoblastoid cell lines, PBMCs, primary monocyte/macrophage cells, and CD4+-T lymphocytes.
The EC
50 values for tenofovir alafenamide were in the range of 2.0 to 14.7 nM. Tenofovir alafenamide displayed antiviral activity in cell culture against all HIV-1 groups (M, N, O), including subtypes A, B, C, D, E, F, and G (EC
50 values ranged from 0.10 to 12.0 nM) and showed activity against HIV-2 (EC
50 values ranged from 0.91 to 2.63 nM).
Resistance: Considering all of the available
in vitro data and data generated in treatment-naïve patients, the following resistance-associated mutations in HIV-1 RT, when present at baseline, may affect the activity of Odefsey: K65R, K70E, K101E, K101P, E138A, E138G, E138K, E138Q, E138R, V179L, Y181C, Y181I, Y181V, M184I, M184V, Y188L, H221Y, F227C, M230I, M230L and the combination of L100I and K103N.
A negative impact by NNRTI mutations other than those listed previously (e.g., mutations K103N or L100I as single mutations) cannot be excluded, since this was not studied
in vivo in a sufficient number of patients.
As with other antiretroviral medicinal products, resistance testing and/or historical resistance data should guide the use of Odefsey (see Precautions).
In vitro: Reduced susceptibility to emtricitabine is associated with M184V/I mutations in HIV-1 RT.
Rilpivirine-resistant strains were selected in cell culture starting from wild-type HIV-1 of different origins and subtypes as well as NNRTI-resistant HIV-1. The most commonly observed amino acid substitutions that emerged included: L100I, K101E, V108I, E138K, V179F, Y181C, H221Y, F227C, and M230I.
HIV-1 isolates with reduced susceptibility to tenofovir alafenamide expressed a K65R mutation in HIV-1 RT; in addition, a K70E mutation in HIV-1 RT has been transiently observed.
In treatment-naïve adult patients: In the Week 144 pooled analysis of antiretroviral-naïve patients receiving elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide (E/C/F/TAF) in the Phase 3 studies GS-US-292-0104 and GS-US-292-0111, the development of one or more primary resistance-associated mutations was observed in HIV-1 isolates from 12 of 866 (1.4%) patients treated with E/C/F/TAF. Among these 12 HIV-1 isolates, the mutations that emerged were M184V/I (n = 11) and K65R/N (n = 2) in RT and T66T/A/I/V (n = 2), E92Q (n = 4), Q148Q/R (n = 1), and N155H (n = 2) in integrase.
In the Week 96 pooled analysis for patients receiving emtricitabine/tenofovir disoproxil fumarate (FTC/TDF) + rilpivirine hydrochloride in the Phase 3 clinical studies TMC278-C209 and TMC278-C215, HIV-1 isolates from 43 patients had an amino acid substitution associated with NNRTI (n = 39) or NRTI (n = 41) resistance. The NNRTI resistance-associated mutations that developed most commonly were: V90I, K101E, E138K/Q, V179I, Y181C, V189I, H221Y and F227C. The presence of V90I and V189I at baseline did not affect the response. Fifty-two percent of HIV-1 isolates with emergent resistance in the rilpivirine arm developed concomitant NNRTI and NRTI mutations, most frequently E138K and M184V. The mutations associated with NRTI resistance that developed in 3 or more patient isolates were: K65R, K70E, M184V/I and K219E.
Through Week 96, fewer patients in the rilpivirine arm with baseline viral load ≤ 100,000 copies/mL had emerging resistance-associated substitutions and/or phenotypic resistance to rilpivirine (7/288) than patients with baseline viral load > 100,000 copies/mL (30/262).
In virologically suppressed patients: One patient with emergent resistance (M184M/I) was identified in a clinical study of virologically suppressed patients who switched from a regimen containing emtricitabine + tenofovir disoproxil fumarate to E/C/F/TAF in a fixed-dose combination (FDC) tablet (GS-US-292-0109, n = 959).
Through Week 96, in patients who switched to Odefsey from emtricitabine/rilpivirine/tenofovir disoproxil fumarate (FTC/RPV/TDF) or efavirenz/emtricitabine/tenofovir disoproxil fumarate (EFV/FTC/TDF) (Studies GS-US-366-1216 and GS-US-366-1160; n = 754), no treatment-emergent resistance-associated mutations were detected.
In patients co-infected with HIV and HBV: In a clinical study of HIV virologically suppressed patients co-infected with chronic hepatitis B, who received E/C/F/TAF for 48 weeks (GS-US-292-1249, n = 72), 2 patients qualified for resistance analysis. In these 2 patients, no amino acid substitutions associated with resistance to any of the components of E/C/F/TAF were identified in HIV-1 or HBV.
Cross-resistance: Emtricitabine-resistant viruses with the M184V/I substitution were cross-resistant to lamivudine, but retained sensitivity to didanosine, stavudine, tenofovir, and zidovudine.
In a panel of 67 HIV-1 recombinant laboratory strains with one resistance-associated mutation at RT positions associated with NNRTI resistance, the only single resistance-associated mutations associated with a loss of susceptibility to rilpivirine were K101P and Y181V/I. The K103N substitution alone did not result in reduced susceptibility to rilpivirine, but the combination of K103N and L100I resulted in a 7-fold reduced susceptibility to rilpivirine. In another study, the Y188L substitution resulted in a reduced susceptibility to rilpivirine of 9-fold for clinical isolates and 6-fold for site-directed mutants.
In patients receiving rilpivirine hydrochloride in combination with FTC/TDF in Phase 3 studies (TMC278-C209 and TMC278-C215 pooled data), most HIV-1 isolates with emergent phenotypic resistance to rilpivirine had cross-resistance to at least one other NNRTI (28/31).
The K65R and also the K70E substitution result in reduced susceptibility to abacavir, didanosine, lamivudine, emtricitabine, and tenofovir, but retain sensitivity to zidovudine.
Clinical data: Clinical efficacy of Odefsey was established from studies conducted with emtricitabine + tenofovir alafenamide when given with elvitegravir + cobicistat as an E/C/F/TAF FDC tablet, from studies conducted with rilpivirine when given with FTC/TDF as individual components or as a FTC/RPV/TDF FDC tablet, and from studies conducted with Odefsey.
Emtricitabine + tenofovir alafenamide containing regimens: Treatment-naïve and virologically suppressed HIV-1 infected adult patients: In Study GS-US-292-0104 and Study GS-US-292-0111, patients received either E/C/F/TAF (n = 866) or elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate (E/C/F/TDF) (n = 867) once daily, both given as FDC tablets.
The mean age was 36 years (range 18-76), 85% were male, 57% were White, 25% were Black, and 10% were Asian. The mean baseline plasma HIV-1 RNA was 4.5 log
10 copies/mL (range 1.3-7.0) and 23% of patients had baseline viral loads > 100,000 copies/mL. The mean baseline CD4+ cell count was 427 cells/mm
3 (range 0-1,360) and 13% had CD4+ cell counts < 200 cells/mm
3.
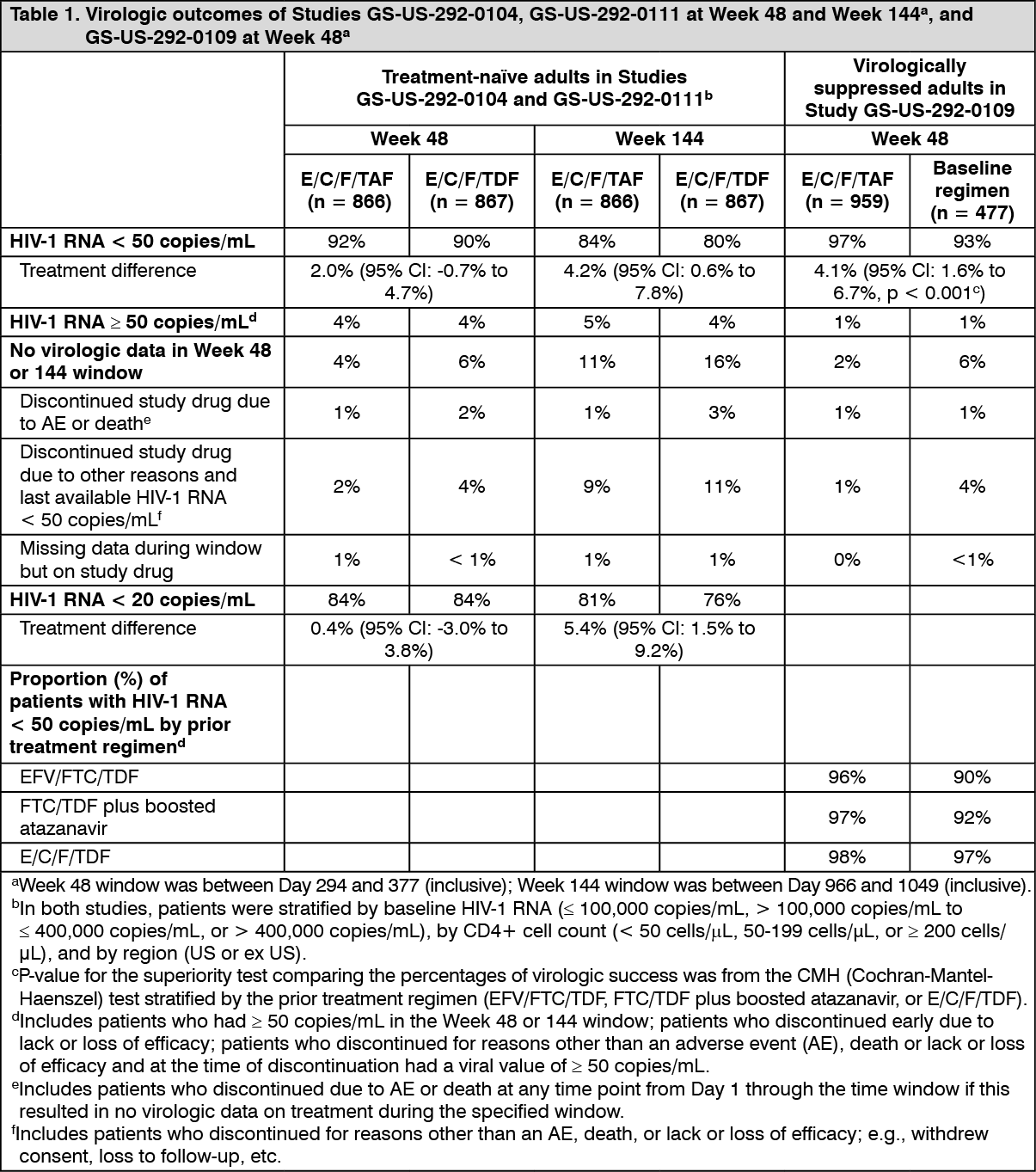
In Studies GS-US-292-0104 and GS-US-292-0111, E/C/F/TAF demonstrated statistical superiority in achieving HIV-1 RNA < 50 copies/mL when compared to E/C/F/TDF at Week 144. The difference in percentage was 4.2% (95% CI: 0.6% to 7.8%). Pooled treatment outcomes at 48 and 144 weeks are shown in Table 1.
In Study GS-US-292-0109, the efficacy and safety of switching from either EFV/FTC/TDF, FTC/TDF plus atazanavir (boosted by either cobicistat or ritonavir), or E/C/F/TDF to E/C/F/TAF FDC tablet were evaluated in a randomised, open-label study of virologically suppressed (HIV-1 RNA < 50 copies/mL) HIV-1 infected adults (n = 959 switching to E/C/F/TAF, n = 477 Stayed on Baseline Regimen [SBR]). Patients had a mean age of 41 years (range 21-77), 89% were male, 67% were White, and 19% were Black. The mean baseline CD4+ cell count was 697 cells/mm
3 (range 79-1,951).
In Study GS-US-292-0109, switching from a tenofovir disoproxil fumarate-based regimen to E/C/F/TAF was superior in maintaining HIV-1 RNA < 50 copies/mL compared to staying on the baseline regimen. Pooled treatment outcomes at 48 weeks are shown in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In Studies GS-US-292-0104 and GS-US-292-0111, the rate of virologic success was similar across patient subgroups (age, gender, race, baseline HIV-1 RNA, or baseline CD4+ cell count).
The mean increase from baseline in CD4+ cell count was 230 cells/mm
3 in E/C/F/TAF-treated patients and 211 cells/mm
3 in E/C/F/TDF-treated patients (p = 0.024) at Week 48 and 326 cells/mm
3 in E/C/F/TAF-treated patients and 305 cells/mm
3 in E/C/F/TDF-treated patients (p = 0.06) at Week 144.
Rilpivirine-containing regimens: Treatment-naïve HIV-1 infected adult patients: The efficacy of rilpivirine is based on the analyses of 96 weeks data from two randomised, double-blind, controlled studies in treatment-naïve patients (TMC278-C209 and emtricitabine + tenofovir disoproxil fumarate subset of TMC278-C215).
In the pooled analysis for TMC278-C209 and TMC278-C215 of 1096 patients who received a background regimen (BR) of FTC/TDF, demographic and baseline characteristics were balanced between the rilpivirine and efavirenz (EFV) arms. The median age was 36 years, 78% were male and 62% White and 24% Black/African American. Median plasma HIV-1 RNA was 5.0 log
10 copies/mL and median CD4+ cell count was 255 cells/mm
3.
Overall response and a subgroup analysis of the virologic response (< 50 HIV-1 RNA copies/mL) at both 48 weeks and 96 weeks, and virologic failure by baseline viral load (pooled data from the two Phase 3 clinical studies, TMC278-C209 and TMC278-C215, for patients receiving the FTC/TDF BR) are presented in Table 2. (See Table 2.)
Click on icon to see table/diagram/image
FTC/TDF + rilpivirine hydrochloride was non-inferior in achieving HIV-1 RNA < 50 copies/mL compared to FTC/TDF + efavirenz.
Odefsey regimen: Virologically suppressed HIV-1 infected adult patients: In Study GS-US-366-1216, the efficacy and safety of switching from FTC/RPV/TDF to Odefsey were evaluated in a randomised, double-blind study of virologically suppressed HIV-1 infected adults. Patients had a mean age of 45 years (range 23-72), 90% were male, 75% were White, and 19% were Black. The mean baseline CD4+ cell count was 709 cells/mm
3 (range: 104-2,527).
In Study GS-US-366-1160, the efficacy and safety of switching from EFV/FTC/TDF to Odefsey were evaluated in a randomised, double-blind study of virologically suppressed HIV-1 infected adults. Patients had a mean age of 48 years (range 19-76), 87% were male, 67% were White, and 27% were Black. The mean baseline CD4+ cell count was 700 cells/mm
3 (range 140-1,862).
Treatment outcomes of Studies GS-US-366-1216 and GS-US-366-1160 are presented Table 3. (See Table 3.)
Click on icon to see table/diagram/image
At Week 96, switching to Odefsey was noninferior in maintaining HIV-1 RNA < 50 copies/mL when compared to patients who stayed on FTC/RPV/TDF or on EFV/FTC/TDF in respective studies.
In Study GS-US-366-1216, the mean change from baseline in CD4+ cell count at Week 96 was 12 cells/mm
3 in patients who switched to Odefsey and 16 cells/mm
3 in those who remained on FTC/RPV/TDF. In Study GS-US-366-1160, the mean change from baseline in CD4+ cell count at Week 96 was 12 cells/mm
3 in patients who switched to Odefsey and 6 cells/mm
3 in those who stayed on EFV/FTC/TDF.
HIV-1 infected adult patients with mild to moderate renal impairment: In Study GS-US-292-0112, the efficacy and safety of E/C/F/TAF FDC tablet were evaluated in an open-label clinical study of 242 HIV-1 infected, virologically suppressed patients with mild to moderate renal impairment (eGFR
CG: 30-69 mL/min).
The mean age was 58 years (range 24-82), with 63 patients (26%) who were ≥ 65 years of age. Seventy-nine percent were male, 63% were White, 18% were Black, and 14% were Asian. Thirty-five percent of patients were on a treatment regimen that did not contain tenofovir disoproxil fumarate. At baseline, median eGFR
CG was 56 mL/min, and 33% of patients had an eGFR
CG from 30 to 49 mL/min. The mean baseline CD4+ cell count was 664 cells/mm
3 (range 126-1,813).
At Week 144, 83.1% (197/237 patients) maintained HIV-1 RNA < 50 copies/mL after switching to E/C/F/TAF FDC tablet.
In Study GS-US-292-1825, the efficacy and safety of E/C/F/TAF were evaluated in a single arm, open-label clinical study in which 55 HIV-1 infected adults with end stage renal disease (eGFRCG < 15 mL/min) on chronic haemodialysis for at least 6 months before switching to E/C/F/TAF FDC tablet. Patients were virologically suppressed (HIV-1 RNA < 50 copies/mL) for at least 6 months before switching.
The mean age was 48 years (range 23-64). Seventy-six percent were male, 82% were Black and 18% were White. Fifteen percent of patients identified as Hispanic/Latino. The mean baseline CD4+ cell count was 545 cells/mm3 (range 205-1473). At Week 48, 81.8% (45/55 patients) maintained HIV-1 RNA < 50 copies/mL after switching to E/C/F/TAF. There were no clinically significant changes in fasting lipid laboratory tests in patients who switched.
Patients co-infected with HIV and HBV: In open-label Study GS-US-292-1249, the efficacy and safety of E/C/F/TAF were evaluated in adult patients co-infected with HIV-1 and chronic hepatitis B. Sixty-nine of the 72 patients were on prior TDF-containing antiretroviral therapy. At the start of treatment with E/C/F/TAF, the 72 patients had been HIV-suppressed (HIV-1 RNA < 50 copies/mL) for at least 6 months with or without suppression of HBV DNA and had compensated liver function. The mean age was 50 years (range 28-67), 92% of patients were male, 69% were White, 18% were Black, and 10% were Asian. The mean baseline CD4+ cell count was 636 cells/mm
3 (range 263-1498). Eighty-six percent of patients (62/72) were HBV suppressed (HBV DNA < 29 IU/mL) and 42% (30/72) were HBeAg positive at baseline.
Of the patients who were HBeAg positive at baseline, 1/30 (3.3%) achieved seroconversion to anti-HBe at Week 48. Of the patients who were HBsAg positive at baseline, 3/70 (4.3%) achieved seroconversion to anti-HBs at Week 48.
At Week 48, 92% of patients (66/72) maintained HIV-1 RNA < 50 copies/mL after switching to E/C/F/TAF. The mean change from baseline in CD4+ cell count at Week 48 was -2 cells/mm
3. Ninety-two percent (66/72 patients) had HBV DNA < 29 IU/mL using missing = failure analysis at Week 48. Of the 62 patients who were HBV suppressed at baseline, 59 remained suppressed and 3 had missing data. Of the 10 patients who were not HBV suppressed at baseline (HBV DNA ≥ 29 IU/mL), 7 became suppressed, 2 remained detectable, and 1 had missing data. Alanine aminotransferase (ALT) normalisation was achieved in 40% (4/10) of subjects with ALT greater than upper limit of normal (ULN) at baseline.
There are limited clinical data on the use of E/C/F/TAF in HIV/HBV co-infected patients who are treatment-naïve.
Changes in measures of bone mineral density: In studies in treatment-naïve adult patients, E/C/F/TAF was associated with smaller reductions in bone mineral density (BMD) compared to E/C/F/TDF through 144 weeks of treatment as measured by dual energy X ray absorptiometry (DXA) analysis of hip (mean change: -0.8%
versus -3.4%, p < 0.001) and lumbar spine (mean change: -0.9%
versus -3.0%, p < 0.001).
Small improvements in BMD were noted at 48 weeks after switching to E/C/F/TAF compared to maintaining the tenofovir disoproxil fumarate-containing regimen.
In Odefsey studies in virologically suppressed adult patients, increases in BMD were noted at 96 weeks after switching to Odefsey compared to minimal changes with maintaining FTC/RPV/TDF or EFV/FTC/TDF at the hip (mean change 1.6% for Odefsey
versus -0.6% for FTC/RPV/TDF, p<0.001; 1.8% for Odefsey
versus -0.6% for EFV/FTC/TDF, p<0.001) and the spine (mean change 2.0% for Odefsey
versus -0.3% for FTC/RPV/TDF, p<0.001; 1.7% for Odefsey
versus 0.1% for EFV/FTC/TDF, p<0.001).
Changes in measures of renal function: In studies in treatment-naïve adult patients, E/C/F/TAF was associated with lower impact on renal safety parameters (as measured after 144 weeks treatment by eGFR
CG and urine protein to creatinine ratio [UPCR] and after 96 weeks treatment by urine albumin to creatinine ratio [UACR]) compared to E/C/F/TDF. Through 144 weeks of treatment, no subject discontinued E/C/F/TAF due to a treatment-emergent renal adverse event compared with 12 subjects who discontinued E/C/F/TDF (p < 0.001). In studies in virologically suppressed adult patients, through 96 weeks of treatment there were minimal changes or decreases in albuminuria (UACR) in patients receiving Odefsey compared with increases from baseline in patients who stayed on FTC/RPV/TDF or EFV/FTC/TDF. See also Precautions.
Paediatric population: Emtricitabine + tenofovir alafenamide regimen: In Study GS-US-292-0106, the efficacy, safety, and pharmacokinetics of E/C/F/TAF FDC tablet were evaluated in an open-label study of 50 HIV-1 infected, treatment-naïve adolescents. Patients had a mean age of 15 years (range 12-17), were 56% female, 12% Asian, and 88% Black. At baseline, median plasma HIV-1 RNA was 4.7 log
10 copies/mL, median CD4+ cell count was 456 cells/mm
3 (range 95 to 1,110), and median CD4+% was 23% (range 7-45). Overall, 22% had baseline plasma HIV-1 RNA > 100,000 copies/mL.
At 48 weeks, 92% (46/50) achieved HIV-1 RNA < 50 copies/mL, similar to response rates in studies of treatment-naïve HIV-1 infected adults. No emergent resistance to E/C/F/TAF was detected through Week 48.
Rilpivirine-containing regimen: The pharmacokinetics, safety, tolerability, and efficacy of rilpivirine 25 mg once daily, in combination with an investigator-selected BR containing two NRTIs, were evaluated in Study TMC278-C213, a single-arm, open-label Phase 2 study in antiretroviral-naïve HIV-1 infected paediatric patients 12 to < 18 years of age and weighing at least 32 kg. The median duration of exposure for patients was 63.5 weeks.
Thirty-six patients had a median age of 14.5 years and were 55.6% female, 88.9% Black, and 11.1% Asian. The median baseline plasma HIV-1 RNA was 4.8 log
10 copies/mL, and the median baseline CD4+ cell count was 414 cells/mm
3. The proportion of patients with HIV-1 RNA < 50 copies/mL at Week 48 (TLOVR) was 72.2% (26/36). The combination of NRTIs most frequently used together with rilpivirine was FTC/TDF (24 subjects [66.7%]).
The proportion of responders was higher in subjects with a baseline viral load ≤ 100,000 copies/mL (78.6%, 22/28) as compared to those with a baseline viral load > 100,000 copies/mL (50.0%, 4/8). The proportion of virologic failures was 22.2% (8/36).
The European Medicines Agency has deferred the obligation to submit the results of studies with Odefsey in one or more subsets of the paediatric population in the treatment of human HIV-1 infection (see Dosage & Administration for information on paediatric use).
Pregnancy: Rilpivirine (one of the components of Odefsey) in combination with a background regimen was evaluated in Study TMC114HIV3015 in 19 pregnant women during the 2
nd and 3
rd trimesters, and postpartum. The pharmacokinetic data demonstrate that total exposure (AUC) to rilpivirine as a part of an antiretroviral regimen was approximately 30% lower during pregnancy compared with postpartum (6-12 weeks). The virologic response was generally preserved throughout the study: of the 12 patients that completed the study, 10 patients were suppressed at the end of the study; in the other 2 patients an increase in viral load was observed only postpartum, for at least 1 patient due to suspected suboptimal adherence. No mother to child transmission occurred in all 10 infants born to the mothers who completed the study and for whom the HIV status was available. Rilpivirine was well tolerated during pregnancy and postpartum. There were no new safety findings compared with the known safety profile of rilpivirine in HIV-1 infected adults (see Dosage & Administration, Precautions and Pharmacokinetics as follows).
Pharmacokinetics: Absorption: Odefsey: Emtricitabine and tenofovir alafenamide exposures were bioequivalent when comparing one Odefsey 200/25/25 mg film-coated tablet to elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide (150/150/200/10 mg) fixed-dose combination tablet following single dose administration to healthy subjects (n = 82) under fed conditions. Rilpivirine exposures were bioequivalent when comparing Odefsey 200/25/25 mg to one rilpivirine (as hydrochloride) 25 mg film-coated tablet following single dose administration to healthy subjects (n = 95) under fed conditions.
Emtricitabine is rapidly and extensively absorbed following oral administration with peak plasma concentrations occurring at 1 to 2 hours post-dose. Following multiple dose oral administration of emtricitabine to 20 HIV-1 infected subjects, the (mean ± SD) area-under the plasma concentration-time curve over a 24-hour dosing interval (AUC) was 10.0 ± 3.1 h·μg/mL. The mean steady-state plasma trough concentration at 24 hours post-dose was equal to or greater than the mean
in vitro IC
90 value for anti-HIV-1 activity. The absolute bioavailability of emtricitabine from 200 mg hard capsules was estimated to be 93%. Emtricitabine systemic exposure was unaffected when emtricitabine was administered with food.
After oral administration, the maximum plasma concentration of rilpivirine is generally achieved within 4 to 5 hours. The absolute bioavailability of rilpivirine is unknown. Relative to fasting conditions, the administration of Odefsey to healthy adult subjects with food resulted in increased rilpivirine exposure (AUC) by 13-72%.
Tenofovir alafenamide is rapidly absorbed following oral administration, with peak plasma concentrations occurring at 15-45 minutes post-dose. Relative to fasting conditions, the administration of Odefsey to healthy adult subjects with food resulted in increased tenofovir alafenamide exposure (AUC) by 45-53%.
It is recommended that Odefsey be taken with food.
Distribution: In vitro binding of emtricitabine to human plasma proteins was < 4% and independent of concentration over the range of 0.02-200 μg/mL.
In vitro binding of rilpivirine to human plasma proteins is approximately 99.7%, primarily to albumin.
In vitro binding of tenofovir to human plasma proteins is < 0.7% and is independent of concentration over the range of 0.01-25 μg/mL.
Ex vivo binding of tenofovir alafenamide to human plasma proteins in samples collected during clinical studies was approximately 80%.
Biotransformation: The biotransformation of emtricitabine includes oxidation of the thiol moiety to form the 3'-sulfoxide diastereomers (approximately 9% of dose) and conjugation with glucuronic acid to form 2'-O-glucuronide (approximately 4% of dose). Emtricitabine did not inhibit
in vitro drug metabolism mediated by any of the major human CYP isoforms involved in drug biotransformation. Also, emtricitabine did not inhibit uridine-5'-diphosphoglucuronyl transferase (UGT), the enzyme responsible for glucuronidation.
In vitro experiments indicate that rilpivirine hydrochloride primarily undergoes oxidative metabolism mediated by the CYP3A system.
Metabolism is a major elimination pathway for tenofovir alafenamide in humans, accounting for > 80% of an oral dose.
In vitro studies have shown that tenofovir alafenamide is metabolised to tenofovir (major metabolite) by cathepsin A in PBMCs (including lymphocytes and other HIV target cells) and macrophages; and by carboxylesterase-1 in hepatocytes.
In vivo, tenofovir alafenamide is hydrolysed within cells to form tenofovir (major metabolite), which is phosphorylated to the active metabolite tenofovir diphosphate. In human clinical studies, a 10 mg oral dose of tenofovir alafenamide given with emtricitabine, cobicistat and elvitegravir resulted in tenofovir diphosphate concentrations > 4-fold higher in PBMCs and > 90% lower concentrations of tenofovir in plasma as compared to a 245 mg oral dose of tenofovir disoproxil (as fumarate) given with emtricitabine, cobicistat and elvitegravir.
In vitro, tenofovir alafenamide is not metabolised by CYP1A2, CYP2C8, CYP2C9, CYP2C19, or CYP2D6. Tenofovir alafenamide is minimally metabolised by CYP3A4. Upon co-administration with the moderate CYP3A inducer probe efavirenz, tenofovir alafenamide exposure was not significantly affected. Following administration of tenofovir alafenamide, plasma [
14C]-radioactivity showed a time-dependent profile, with tenofovir alafenamide as the most abundant species in the initial few hours and uric acid in the remaining period.
Elimination: Emtricitabine is primarily excreted by the kidneys with complete recovery of the dose achieved in urine (approximately 86%) and faeces (approximately 14%). Thirteen percent of the emtricitabine dose was recovered in urine as three metabolites. The systemic clearance of emtricitabine averaged 307 mL/min. Following oral administration, the elimination half-life of emtricitabine is approximately 10 hours.
The terminal elimination half-life of rilpivirine is approximately 45 hours. After single dose oral administration of [
14C]-rilpivirine, on average 85% and 6.1% of the radioactivity could be retrieved in faeces and urine, respectively. In faeces, unchanged rilpivirine accounted for on average 25% of the administered dose. Only trace amounts of unchanged rilpivirine (< 1% of dose) were detected in urine.
Renal excretion of intact tenofovir alafenamide is a minor pathway with < 1% of the dose eliminated in urine. Tenofovir alafenamide is mainly eliminated following metabolism to tenofovir. Tenofovir is renally eliminated by both glomerular filtration and active tubular secretion.
Pharmacokinetics in special populations: Age, gender and ethnicity: No clinically relevant pharmacokinetic differences due to age, gender or ethnicity have been identified for emtricitabine, rilpivirine or tenofovir alafenamide.
Paediatric population: The pharmacokinetics of rilpivirine in antiretroviral-naïve HIV-1 infected paediatric patients 12 to < 18 years of age receiving rilpivirine 25 mg once daily was comparable to that in treatment-naïve HIV-1 infected adults receiving rilpivirine 25 mg once daily. There was no impact of body weight on rilpivirine pharmacokinetics in paediatric patients in Study C213 (33 to 93 kg), similar to what was observed in adults. The pharmacokinetics of rilpivirine in paediatric patients < 12 years of age is under investigation.
Exposures of emtricitabine and tenofovir alafenamide given with elvitegravir + cobicistat achieved in 24 paediatric patients aged 12 to < 18 years were similar to exposures achieved in treatment-naïve adults (Table 4). (See Table 4.)
Click on icon to see table/diagram/image
Renal impairment:
No clinically relevant differences in tenofovir alafenamide or tenofovir pharmacokinetics were observed between healthy subjects and patients with severe renal impairment (estimated CrCl ≥ 15 mL/min and < 30 mL/min) in a Phase 1 study of tenofovir alafenamide. In a separate Phase 1 study of emtricitabine alone, mean systemic emtricitabine exposure was higher in patients with severe renal impairment (estimated CrCl < 30 mL/min) (33.7 μg·h/mL) than in subjects with normal renal function (11.8 μg·h/mL). The safety of emtricitabine + tenofovir alafenamide has not been established in patients with severe renal impairment (estimated CrCl ≥ 15 mL/min and < 30 mL/min).
Exposures of emtricitabine and tenofovir in 12 patients with end stage renal disease (estimated CrCl < 15 mL/min) on chronic haemodialysis who received emtricitabine + tenofovir alafenamide in combination with elvitegravir + cobicistat as a fixed-dose combination tablet (E/C/F/TAF) in Study GS-US-292-1825 were significantly higher than in patients with normal renal function. No clinically relevant differences in tenofovir alafenamide pharmacokinetics were observed in patients with end stage renal disease on chronic haemodialysis as compared to those with normal renal function. There were no new safety issues identified in patients with end stage renal disease on chronic haemodialysis receiving emtricitabine + tenofovir alafenamide, given with elvitegravir + cobicistat as a fixed-dose combination tablet (see Adverse Reactions).
There are no pharmacokinetic data on emtricitabine or tenofovir alafenamide in patients with end stage renal disease (estimated CrCl < 15 mL/min) not on chronic haemodialysis. The safety of emtricitabine and tenofovir alafenamide has not been established in these patients.
The pharmacokinetics of rilpivirine have not been studied in patients with renal insufficiency. Renal elimination of rilpivirine is negligible. In patients with severe renal impairment or end-stage renal disease, plasma concentrations may be increased due to alteration of drug absorption, distribution and/or metabolism secondary to renal dysfunction. As rilpivirine is highly bound to plasma proteins, it is unlikely that it will be significantly removed by haemodialysis or peritoneal dialysis (see Overdosage).
Hepatic impairment: The pharmacokinetics of emtricitabine have not been studied in patients with varying degrees of hepatic insufficiency; however emtricitabine is not significantly metabolised by liver enzymes, so the impact of liver impairment should be limited.
Rilpivirine hydrochloride is primarily metabolised and eliminated by the liver. In a study comparing 8 patients with mild hepatic impairment (Child-Pugh Class A) to 8 matched controls and 8 patients with moderate hepatic impairment (Child-Pugh Class B) to 8 matched controls, the multiple dose exposure of rilpivirine was 47% higher in patients with mild hepatic impairment and 5% higher in patients with moderate hepatic impairment. However, it may not be excluded that the pharmacologically active, unbound, rilpivirine exposure is significantly increased in moderate impairment. Rilpivirine has not been studied in patients with severe hepatic impairment (Child Pugh Class C) (see Dosage & Administration).
Clinically relevant changes in the pharmacokinetics of tenofovir alafenamide or its metabolite tenofovir were not observed in patients with mild or moderate hepatic impairment. In patients with severe hepatic impairment, total plasma concentrations of tenofovir alafenamide and tenofovir are lower than those seen in subjects with normal hepatic function. When corrected for protein binding, unbound (free) plasma concentrations of tenofovir alafenamide in severe hepatic impairment and normal hepatic function are similar.
Hepatitis B and/or hepatitis C virus co-infection: The pharmacokinetics of emtricitabine, rilpivirine and tenofovir alafenamide have not been fully evaluated in patients co-infected with hepatitis B and/or C virus.
Pregnancy and postpartum: After taking rilpivirine 25 mg once daily as part of an antiretroviral regimen, the total exposure of rilpivirine was lower during pregnancy (similar for the 2
nd and 3
rd trimester) compared with postpartum. The decrease in the unbound free fraction of rilpivirine exposure (ie, active) during pregnancy compared to postpartum was less pronounced than for total exposure of rilpivirine.
In women receiving rilpivirine 25 mg once daily during the 2
nd trimester of pregnancy, mean intra-individual values for total rilpivirine C
max, AUC
24h and C
min values were 21%, 29% and 35% lower, respectively, as compared to postpartum; during the 3
rd trimester of pregnancy, C
max, AUC
24h and C
min values were 20%, 31% and 42% lower, respectively, as compared to postpartum.
Toxicology: Preclinical safety data: Non-clinical data on emtricitabine reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction and development.
Non-clinical data on rilpivirine hydrochloride reveal no special hazard for humans based on studies of safety pharmacology, drug disposition, genotoxicity, carcinogenic potential, toxicity to reproduction and development. Liver toxicity associated with liver enzyme induction was observed in rodents. In dogs cholestasis-like effects were noted.
Carcinogenicity studies with rilpivirine in mice and rats revealed tumorigenic potential specific for these species, but are regarded as of no relevance for humans.
Non-clinical studies of tenofovir alafenamide in rats and dogs revealed bone and kidney as the primary target organs of toxicity. Bone toxicity was observed as reduced bone mineral density in rats and dogs at tenofovir exposures at least four times greater than those expected after administration of Odefsey. A minimal infiltration of histiocytes was present in the eye in dogs at tenofovir alafenamide and tenofovir exposures of approximately 4- and 17-times greater, respectively, than those expected after administration of Odefsey.
Tenofovir alafenamide was not mutagenic or clastogenic in conventional genotoxicity assays.
Because there is a lower tenofovir exposure in rats and mice after the administration of tenofovir alafenamide compared to tenofovir disoproxil fumarate, carcinogenicity studies and a rat peri-postnatal study were conducted only with tenofovir disoproxil fumarate. No special hazard for humans was revealed in conventional studies of carcinogenic potential and toxicity to reproduction and development. Reproductive toxicity studies in rats and rabbits showed no effects on mating, fertility, pregnancy or foetal parameters. However, tenofovir disoproxil fumarate reduced the viability index and weight of pups in a peri-postnatal toxicity study at maternally toxic doses.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image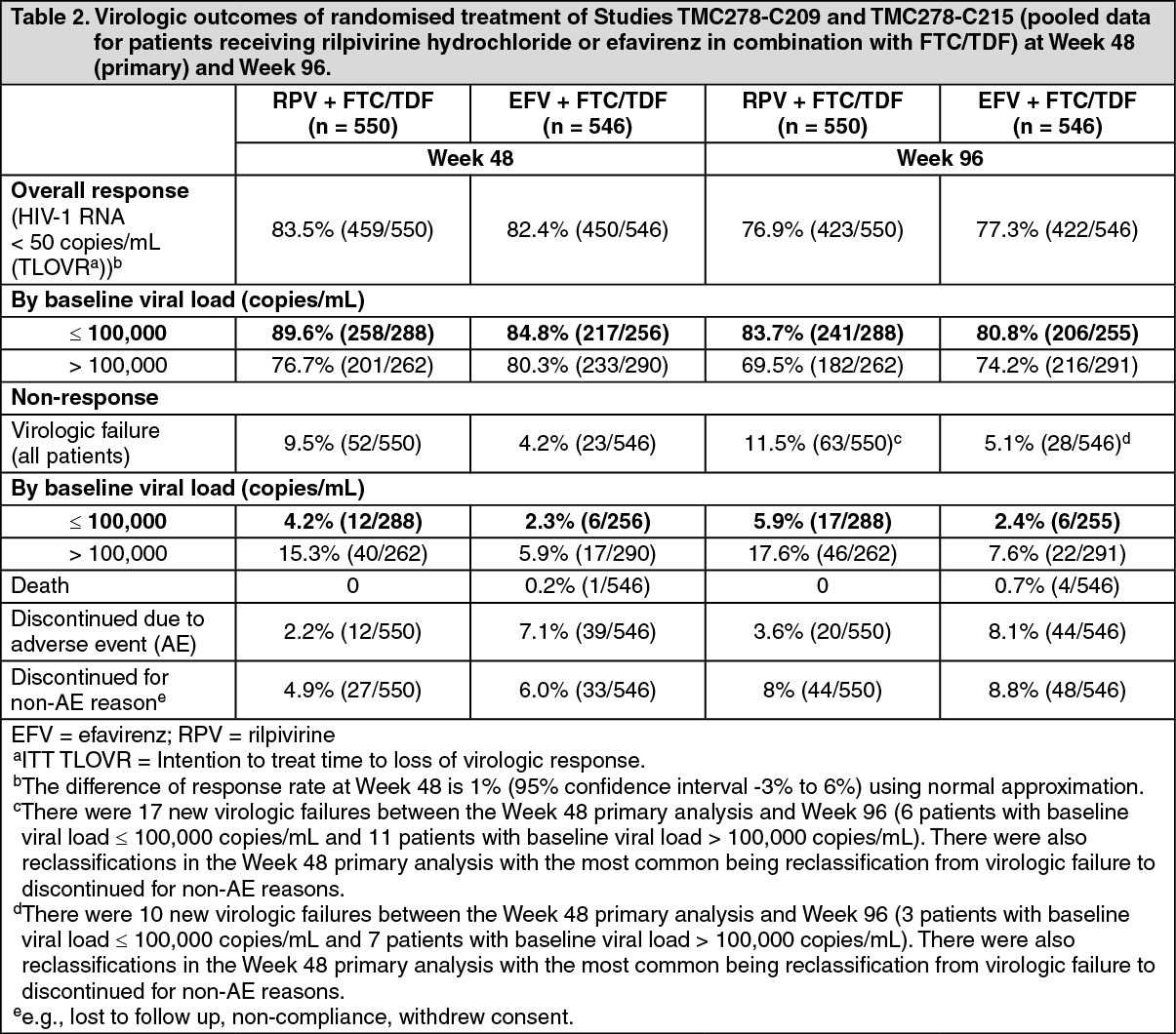 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image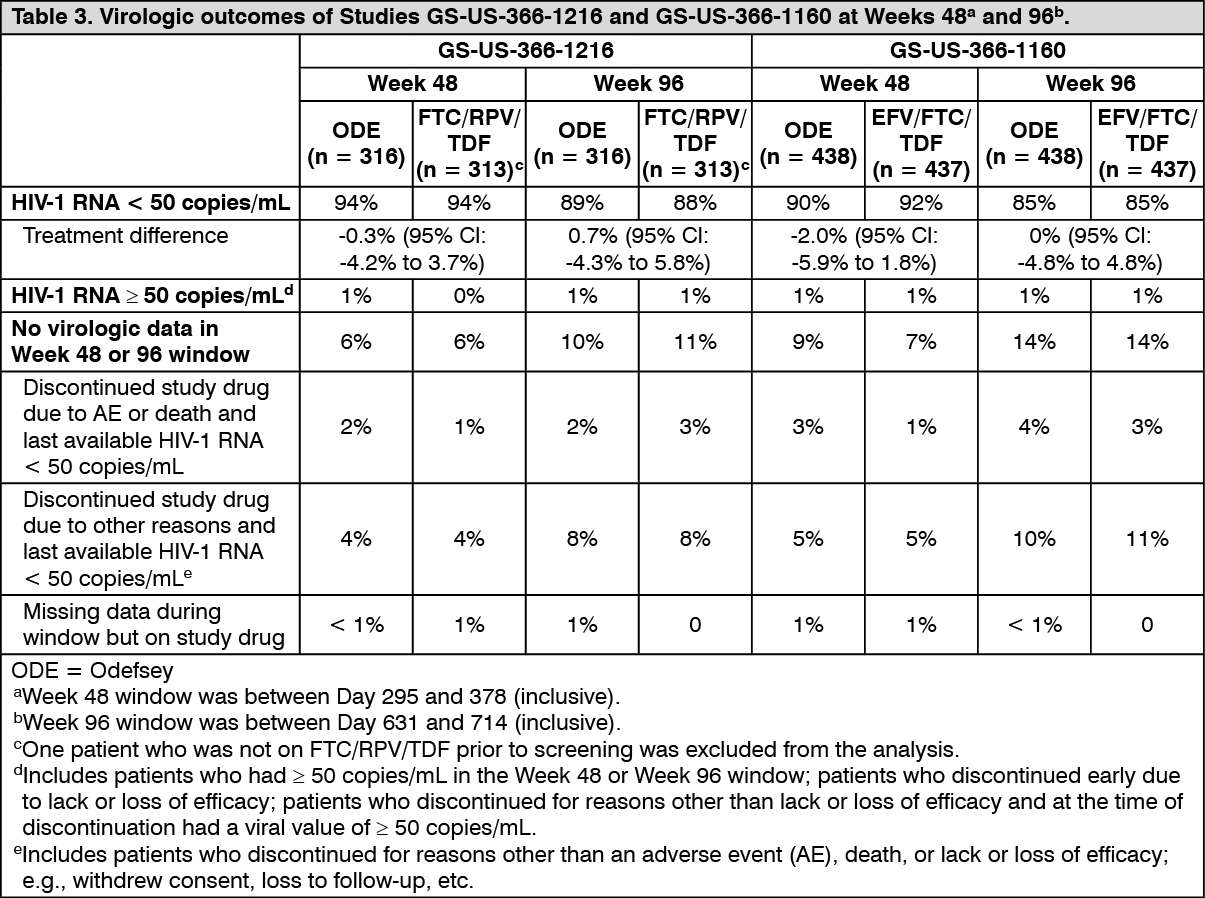 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image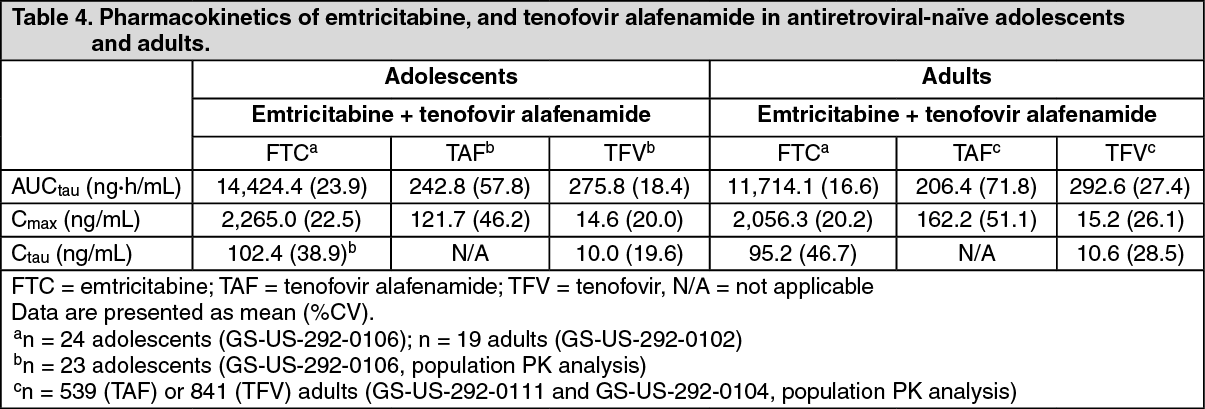 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image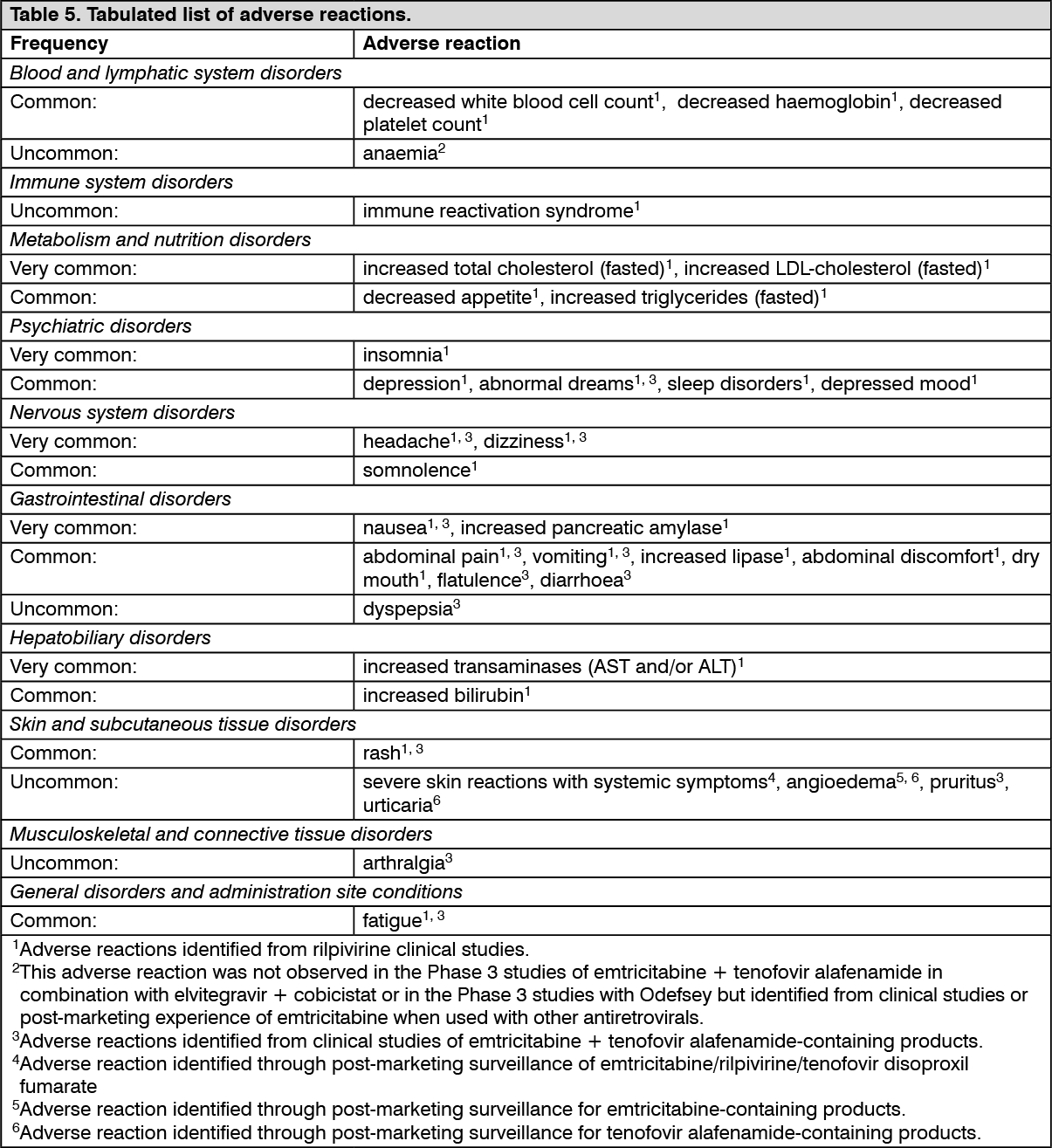 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image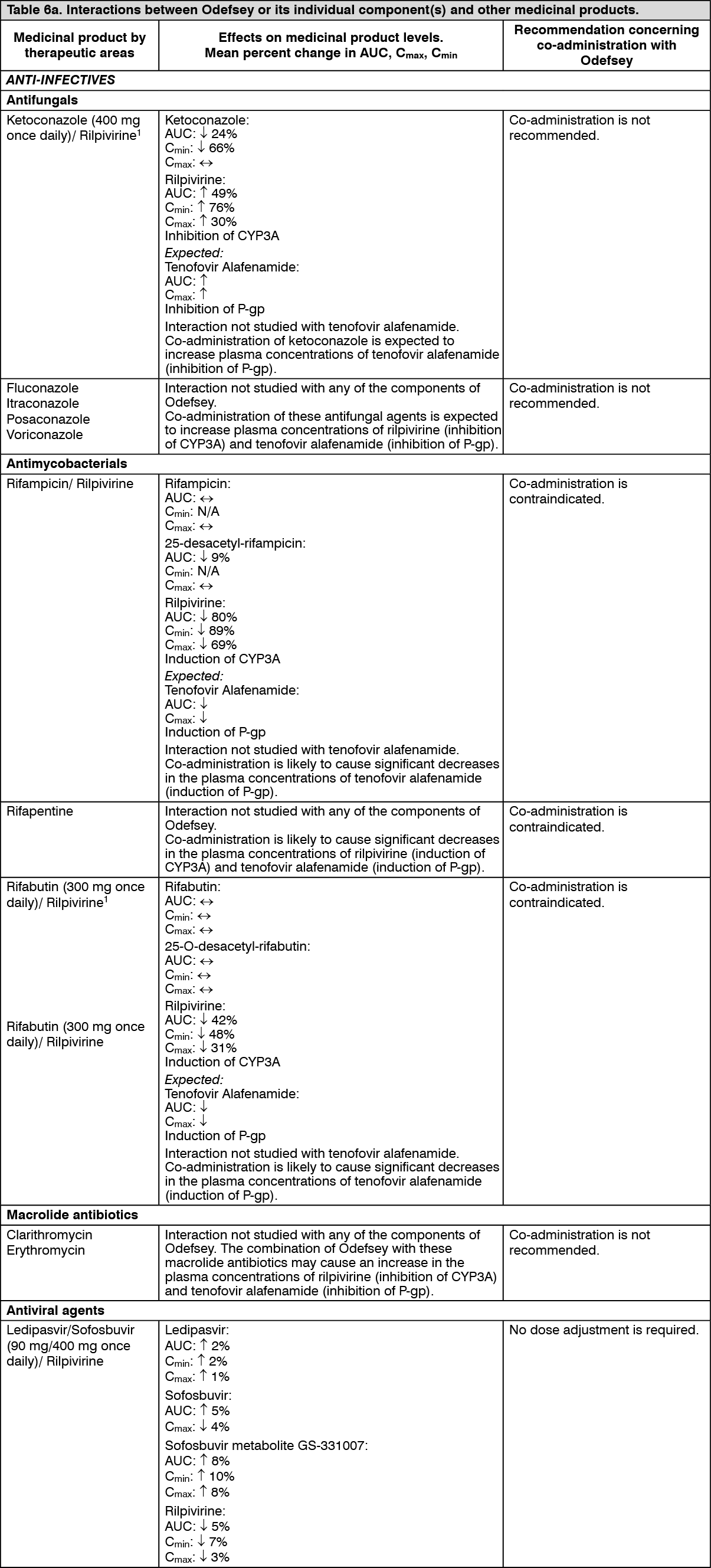 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image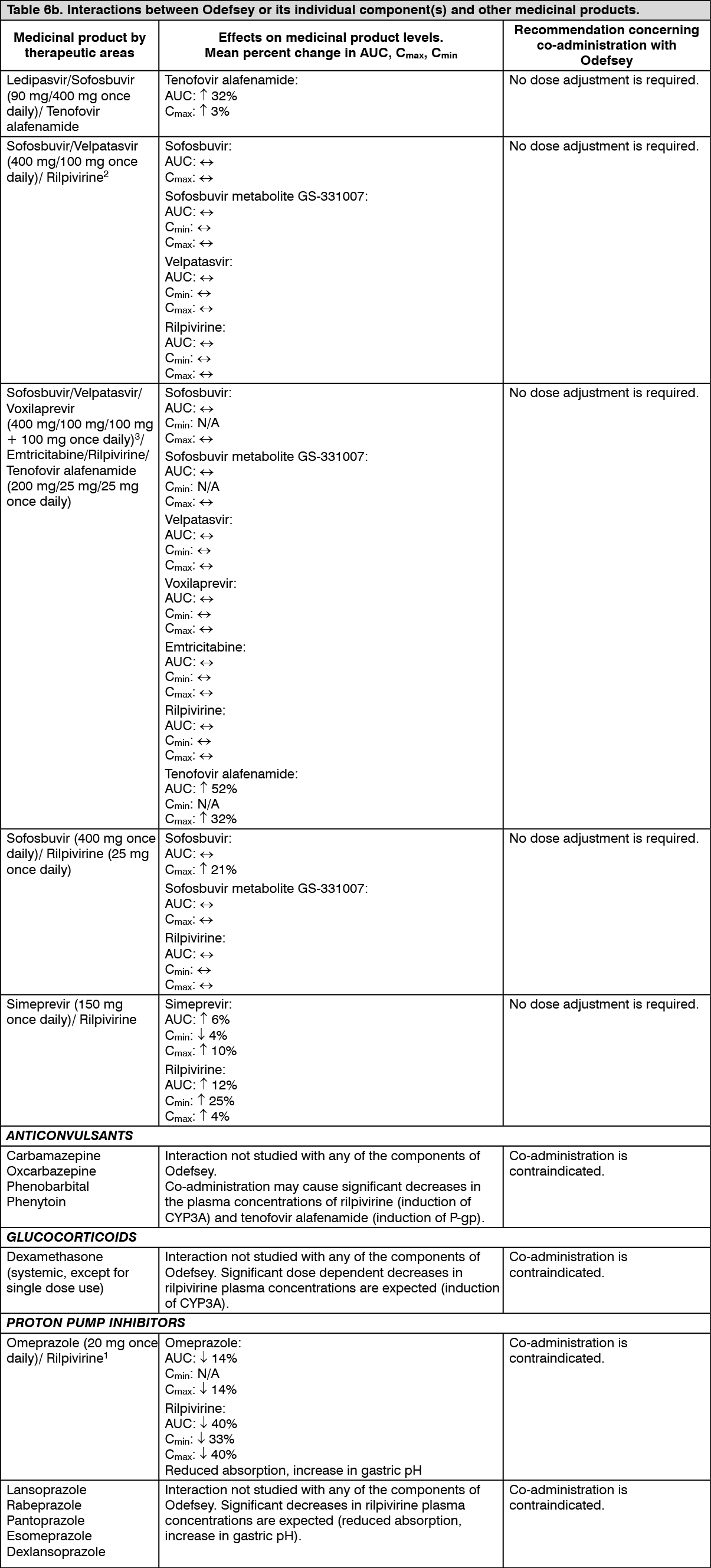 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image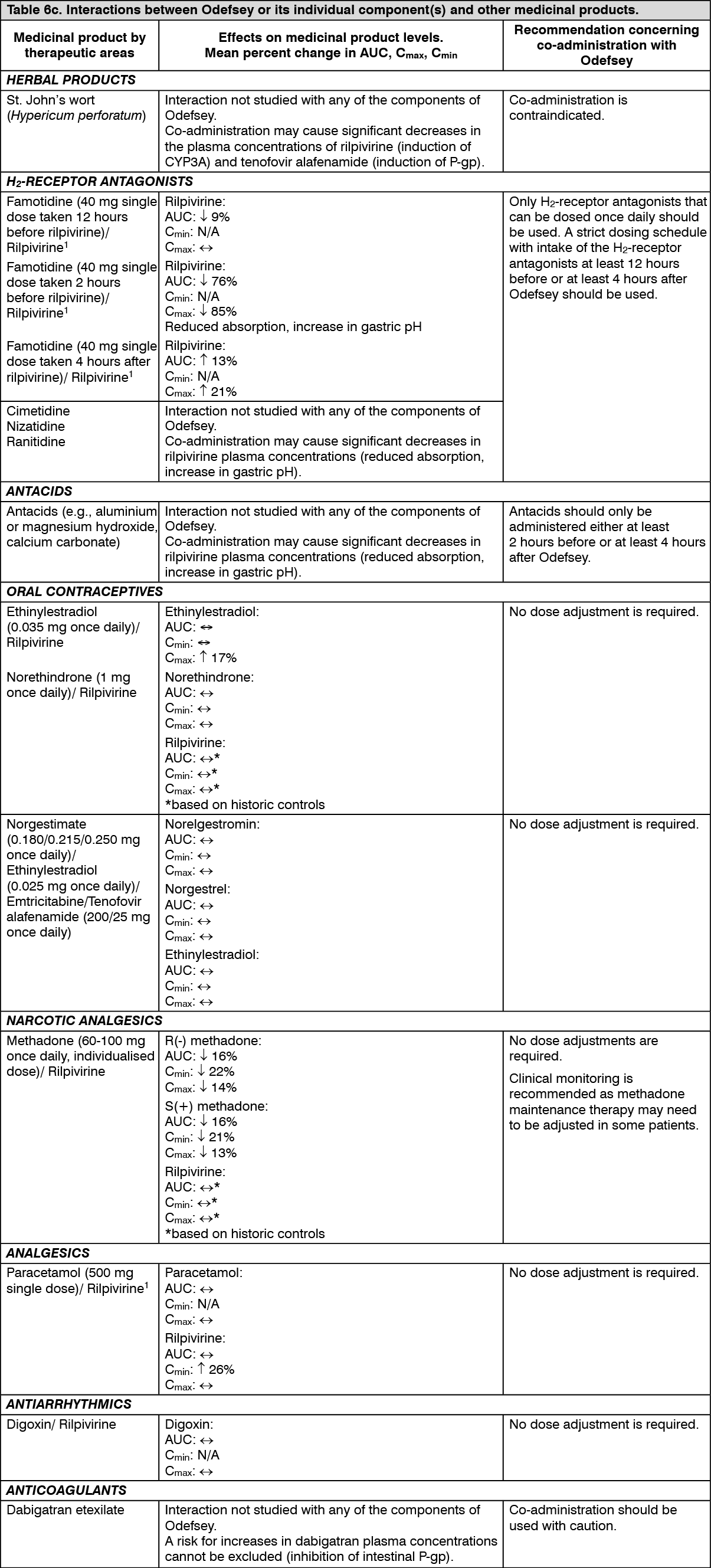 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out