Pharmacotherapeutic group: Antivirals for systemic use, direct acting antivirals.
ATC code: J05AX18.
Pharmacology: Pharmacodynamics: Mechanism of action: Letermovir inhibits the CMV DNA terminase complex which is required for cleavage and packaging of viral progeny DNA. Letermovir affects the formation of proper unit length genomes and interferes with virion maturation.
Antiviral activity: The median EC
50 value of letermovir against a collection of clinical CMV isolates in a cell-culture model of infection was 2.1 nM (range = 0.7 nM to 6.1 nM, n=74).
Viral resistance: In cell culture: The CMV genes UL51, UL56, and UL89 encode subunits of CMV DNA terminase. CMV mutants with reduced susceptibility to letermovir have been confirmed in cell culture. EC
50 values for recombinant CMV mutants expressing the substitutions map to pUL51 (P91S), pUL56 (C25F, S229F, V231A, V231L,V236A, T244K, T244R, L254F, L257F, L257I, F261C, F261L, F261S, Y321C, L328V, M329T, A365S, N368D), and pUL89 (N320H, D344E) were 1.6- to <10-fold higher than those for wild-type reference virus; these substitutions are not likely to be clinically relevant. EC
50 values for recombinant CMV mutants expressing pUL56 substitutions N232Y, V236L, V236M, E237D, E237G, L241P, K258E, C325F, C325R, C325W, C325Y, R369G, R369M, R369S and R369T were 10- to 9,300-fold higher than those for the wild-type reference virus; some of these substitutions have been observed in patients who have experienced prophylaxis failure in clinical studies (see as follows).
In clinical studies: In a Phase 2b trial evaluating letermovir doses of 60, 120, or 240 mg/day or placebo for up to 84 days in 131 HSCT recipients, DNA sequence analysis of a select region of UL56 (amino acids 231 to 369) was performed on samples obtained from 12 letermovir-treated subjects who experienced prophylaxis failure and for whom samples were available for analysis. One subject (who received 60 mg/day) had a letermovir resistant genotypic variant (GV) (V236M).
In a Phase 3 trial (P001), DNA sequence analysis of the entire coding regions of UL56 and UL89 was performed on samples obtained from 40 letermovir-treated subjects in the FAS population who experienced prophylaxis failure and for whom samples were available for analysis. Two subjects had letermovir-resistant GVs detected, both with substitutions mapping to pUL56. One subject had the substitution V236M and the other subject had the substitution E237G. One additional subject, who had detectable CMV DNA at baseline (and was therefore not in the FAS population), had pUL56 substitutions, C325W and R369T, detected after discontinuing letermovir.
Cross-resistance: Cross-resistance is not likely with medicinal products with a different mechanism of action. Letermovir is fully active against viral populations with substitutions conferring resistance to CMV DNA polymerase inhibitors (ganciclovir, cidofovir, and foscarnet). A panel of recombinant CMV strains with substitutions conferring resistance to letermovir was fully susceptible to cidofovir, foscarnet and ganciclovir with the exception of a recombinant strain with the pUL56 E237G substitution which confers a 2.1-fold reduction in ganciclovir susceptibility relative to wild-type.
Cardiac electrophysiology: The effect of letermovir on doses up to 960 mg given IV on the QTc interval was evaluated in a randomised, single-dose, placebo- and active-controlled (moxifloxacin 400 mg oral) 4-period crossover thorough QT trial in 38 healthy subjects. Letermovir does not prolong QTc to any clinically relevant extent following the 960 mg IV dose with plasma concentrations approximately 2-fold higher than the 480 mg IV dose.
Clinical efficacy and safety: Adult CMV-seropositive recipients [R+] of an allogeneic hematopoietic stem cell transplant: To evaluate letermovir prophylaxis as a preventive strategy for CMV infection or disease, the efficacy of letermovir was assessed in a multicenter, double-blind, placebo-controlled Phase 3 trial (P001) in adult CMV-seropositive recipients [R+] of an allogeneic HSCT. Subjects were randomised (2:1) to receive either letermovir at a dose of 480 mg once daily adjusted to 240 mg when co-administered with cyclosporine, or placebo. Randomisation was stratified by investigational site and risk (high vs. low) for CMV reactivation at the time of study entry. Letermovir was initiated after HSCT (Day 0-28 post-transplant) and continued through Week 14 post-transplant. Letermovir was administered either orally or IV; the dose of letermovir was the same regardless of the route of administration. Subjects were monitored through Week 24 post-transplant for the primary efficacy endpoint with continued follow-up through Week 48 post-transplant.
Subjects received CMV DNA monitoring weekly until post-transplant week 14 and then bi-weekly until post-transplant week 24, with initiation of standard-of-care CMV pre-emptive therapy if CMV DNAemia was considered clinically significant. Subjects had continued follow-up through Week 48 post-transplant.
Among the 565 treated subjects, 373 subjects received letermovir (including 99 subjects who received at least one IV dose) and 192 received placebo (including 48 subjects who received at least one IV dose). The median time to starting letermovir was 9 days after transplantation. Thirty-seven percent (37%) of subjects were engrafted at baseline. The median age was 54 years (range: 18 to 78 years); 56 (15.0%) subjects were 65 years of age or older: 58% were male; 82% were White; 10% were Asian; 2% were Black or African; and 7% were Hispanic or Latino. At baseline, 50% of subjects received a myeloablative regimen, 52% were receiving cyclosporine, and 42% were receiving tacrolimus. The most common primary reasons for transplant were acute myeloid leukemia (38%), myeloblastic syndrome (15%), and lymphoma (13%). Twelve percent (12%) of subjects were positive for CMV DNA at baseline.
At baseline, 31% of subjects were at high risk for reactivation as defined by one or more of the following criteria: Human Leukocyte Antigen (HLA)-related (sibling) donor with at least one mismatch at one of the following three HLA-gene loci: HLA-A, -B or -DR, haploidentical donor; unrelated donor with at least one mismatch at one of the following four HLA-gene loci: HLA-A, -B, -C and -DRB1; use of umbilical cord blood as stem cell source; use of
ex vivo T-cell-depleted grafts; Grade 2 or greater Graft-Versus-Host Disease (GVHD), requiring systemic corticosteroids.
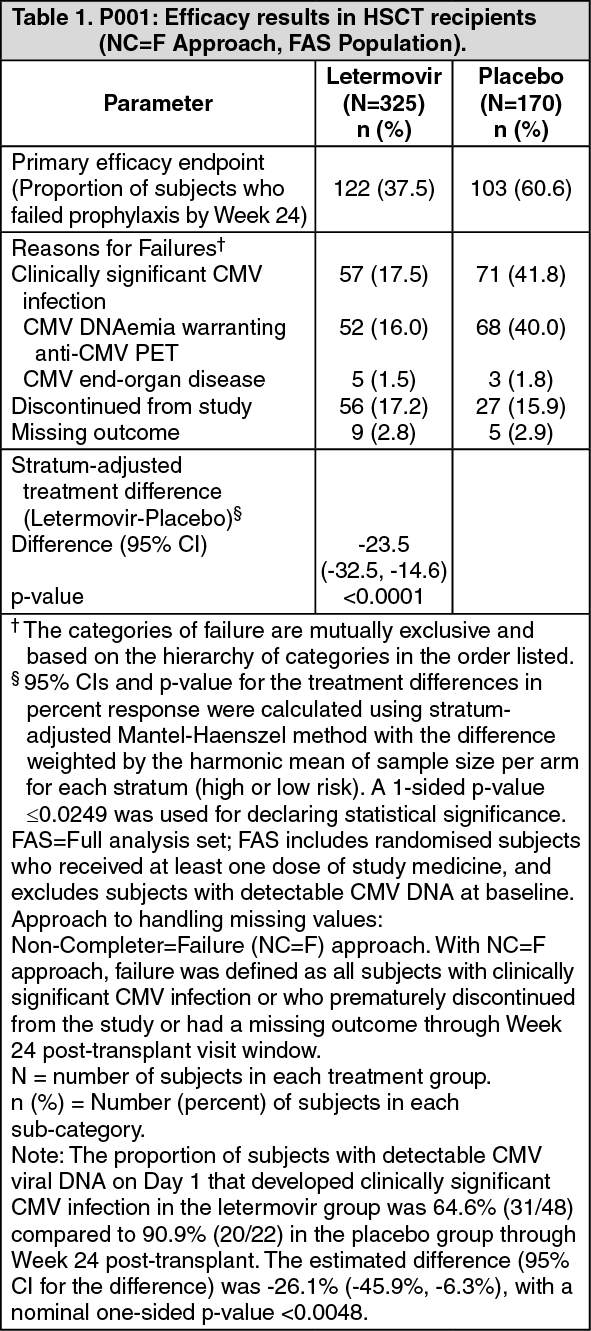
Primary efficacy endpoint: The primary efficacy endpoint of clinically significant CMV infection in P001 was defined by the incidence of CMV DNAemia warranting anti-CMV pre-emptive therapy (PET) or the occurrence of CMV end-organ disease. The Non-Completer=Failure (NC=F) approach was used, where subjects who discontinued from the study prior to Week 24 post-transplant or had a missing outcome at Week 24 post-transplant were counted as failures.
Letermovir demonstrated superior efficacy over placebo in the analysis of the primary endpoint, as shown in Table 1. The estimated treatment difference of -23.5% was statistically significant (one-sided p-value <0.0001). (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Factors associated with CMV DNAemia after Week 14 post-transplant among letermovir-treated subjects included high risk for CMV reactivation at baseline, GVHD, use of corticosteroids, and CMV negative donor serostatus. (See Figure 1.)
Click on icon to see table/diagram/image
There were no differences in the incidence of or time to engraftment between the PREVYMIS and placebo groups.
Efficacy consistently favoured letermovir across subgroups including low and high risk for CMV reactivation, conditioning regimens, and concomitant immunosuppressive regimens (see Figure 2).
Click on icon to see table/diagram/image
Paediatric population: The European Medicines Agency has deferred the obligation to submit the results of studies with PREVYMIS in one or more subsets of the paediatric population for prophylaxis of cytomegalovirus infection (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: The pharmacokinetics of letermovir have been characterized following oral and IV administration in healthy subjects and HSCT recipients. Letermovir exposure increased in a greater than dose-proportional manner with both oral or IV administration. The mechanism is likely saturation/autoinhibition of OATP1B1/3.
In healthy subjects, the geometric mean steady-state AUC and C
max values were 71,500 ng·hr/mL and 13,000 ng/mL, respectively, with 480 mg once daily oral letermovir.
Letermovir reached steady-state in 9 to 10 days with an accumulation ratio of 1.2 for AUC and 1.0 for C
max.
In HSCT recipients, letermovir AUC was estimated using population pharmacokinetic analyses using Phase 3 data (see Table 2). Differences in exposure across treatment regimens are not clinically relevant; efficacy was consistent across the range of exposures observed in P001. (See Table 2.)
Click on icon to see table/diagram/image
Absorption: Letermovir was absorbed rapidly with a median time to maximum plasma concentration (T
max) of 1.5 to 3.0 hours and declined in a biphasic manner. In HSCT recipients, bioavailability of letermovir was estimated to be approximately 35% with 480 mg once daily oral letermovir administered without cyclosporine. The inter-individual variability for bioavailability was estimated to be approximately 37%.
Effect of cyclosporine: In HSCT recipients, co-administration of cyclosporine increased plasma concentrations of letermovir due to inhibition of OATP1B. Bioavailability of letermovir was estimated to be approximately 85% with 240 mg once daily oral letermovir co-administered with cyclosporine in patients.
If letermovir is co-administered with cyclosporine, the recommended dose of letermovir is 240 mg once daily (see Dosage & Administration).
Effect of food: In healthy subjects, oral administration of 480 mg single dose of letermovir with a standard high fat and high calorie meal did not have any effect on the overall exposure (AUC) and resulted in approximately 30% increase in peak levels (C
max) of letermovir. Letermovir may be administered orally with or without food as has been done in the clinical studies (see Dosage & Administration).
Distribution: Based on population pharmacokinetic analyses, the mean steady-state volume of distribution is estimated to be 45.5 L following intravenous administration in HSCT recipients.
Letermovir is extensively bound (98.2%) to human plasma proteins, independent of the concentration range (3 to 100 mg/L) evaluated,
in vitro. Some saturation was observed at lower concentrations. Blood to plasma partitioning of letermovir is 0.56 and independent of the concentration range (0.1 to 10 mg/L) evaluated
in vitro.
In preclinical distribution studies, letermovir is distributed to organs and tissues with the highest concentrations observed in the gastrointestinal tract, bile duct and liver and low concentrations in the brain.
Biotransformation: The majority of letermovir-related components in plasma is unchanged parent (96.6%). No major metabolites are detected in plasma. Letermovir is partly eliminated by glucuronidation mediated by UGT1A1/1A3.
Elimination: The mean apparent terminal half-life for letermovir is approximately 12 hours with 480 mg IV letermovir in healthy subjects. The major elimination pathways of letermovir is biliary excretion as well as direct glucuronidation. The process involves the hepatic uptake transporters OATP1B1 and 3 followed by UGT1A1/3 catalysed glucuronidation.
Based on population pharmacokinetic analyses, letermovir steady-state apparent CL is estimated to be 4.84 L/hr following intravenous administration of 480 mg in HSCT recipients. The inter-individual variability for CL is estimated to be 24.6%.
Excretion: After oral administration of radio-labeled letermovir, 93.3% of radioactivity was recovered in faeces. The majority of letermovir was biliary excreted as unchanged parent with a minor amount (6% of dose) as an acyl-glucuronide metabolite in faeces. The acyl-glucuronide is unstable in faeces. Urinary excretion of letermovir was negligible (<2% of dose).
Pharmacokinetics in special populations: Hepatic impairment: Letermovir unbound AUC was approximately 81%- and 4-fold higher in subjects with moderate (Child-Pugh Class B [CP-B], score of 7-9) and severe (Child-Pugh Class C [CP-C], score of 10-15) hepatic impairment, respectively, compared to healthy subjects. The changes in letermovir exposure in subjects with moderate hepatic impairment are not clinically relevant.
Marked increases in letermovir unbound exposure are anticipated in patients with moderate hepatic impairment combined with moderate or severe renal impairment (see Dosage & Administration).
Renal impairment: Letermovir unbound AUC was approximately 115- and 81% higher in subjects with moderate (eGFR of 31.0 to 56.8 mL/min/1.73m
2) and severe (eGFR of 11.9 to 28.1 mL/min/1.73m
2) renal impairment, respectively, compared to healthy subjects. The changes in letermovir exposure due to moderate or severe renal impairment are not considered to be clinically relevant. Subjects with ESRD have not been studied.
Weight: Based on population pharmacokinetic analyses, letermovir AUC is estimated to be 18.7% lower in subjects weighing 80-100 kg compared to subjects weighing 67 kg. This difference is not clinically relevant.
Race: Based on population pharmacokinetic analyses, letermovir AUC is estimated to be 33.2% higher in Asians compared to Whites. This change is not clinically relevant.
Gender: Based on population pharmacokinetic analyses, there is no difference in letermovir pharmacokinetics in females compared to males.
Elderly: Based on population pharmacokinetic analyses, there is no effect of age on letermovir pharmacokinetics. No dose adjustment is required based on age.
Toxicology: Preclinical safety data: General toxicity: Irreversible testicular toxicity was noted only in rats at systemic exposures (AUC) ≥3-fold the exposures in humans at the recommended human dose (RHD). This toxicity was characterized by seminiferous tubular degeneration, and oligospermia and cell debris in the epididymides, with decreased testicular and epididymides weights. There was no testicular toxicity in rats at exposures (AUC) similar to the exposures in humans at the RHD. Testicular toxicity was not observed in mice and monkeys at the highest doses tested at exposures up to 4-fold and 2-fold, respectively, the exposures in humans at the RHD. The relevance to humans is unknown.
It is known that hydroxypropylbetadex can cause kidney vacuolation in rats when given intravenously at doses greater than 50 mg/kg/day. Vacuolation was noted in the kidneys of rats administered IV letermovir formulated with 1500 mg/kg/day of the cyclodextrin excipient hydroxypropylbetadex.
Carcinogenesis: Carcinogenicity studies with letermovir have not been conducted.
Mutagenesis: Letermovir was not genotoxic in a battery of
in vitro or
in vivo assays, including microbial mutagenesis assays, chromosomal aberration in Chinese Hamster Ovary cells, and in an
in vivo mouse micronucleus study.
Reproduction: Fertility: In the fertility and early embryonic development studies in the rat, there were no effects of letermovir on female fertility. In male rats, reduced sperm concentration, reduced sperm motility, and decreased fertility were observed at systemic exposures ≥ 3-fold the AUC in humans at the RHD (see General toxicity as previously mentioned).
In monkeys administered letermovir, there was no evidence of testicular toxicity based on histopathologic evaluation, measurement of testicular size, blood hormone analysis (follicle stimulating hormone, inhibin B and testosterone) and sperm evaluation (sperm count, motility and morphology) at systemic exposures approximately 2-fold the AUC in humans at the RHD.
Development: In rats, maternal toxicity (including decrease in body weight gain) was noted at 250 mg/kg/day (approximately 11-fold the AUC at the RHD); in the offspring, decreased foetal weight with delayed ossification, slightly oedematous foetuses, and increased incidence of shortened umbilical cords and of variations and malformations in the vertebrae, ribs, and pelvis were observed. No maternal or developmental effects were noted at the dose of 50 mg/kg/day (approximately 2.5-fold the AUC at the RHD).
In rabbits, maternal toxicity (including mortality and abortions) was noted at 225 mg/kg/day (approximately 2-fold the AUC at the RHD); in the offspring, an increased incidence of malformations and variations in the vertebrae and ribs were observed.
In the pre- and post-natal developmental study, letermovir was administered orally to pregnant rats. There was no developmental toxicity observed up to the highest exposure tested (2-fold the AUC at the RHD).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image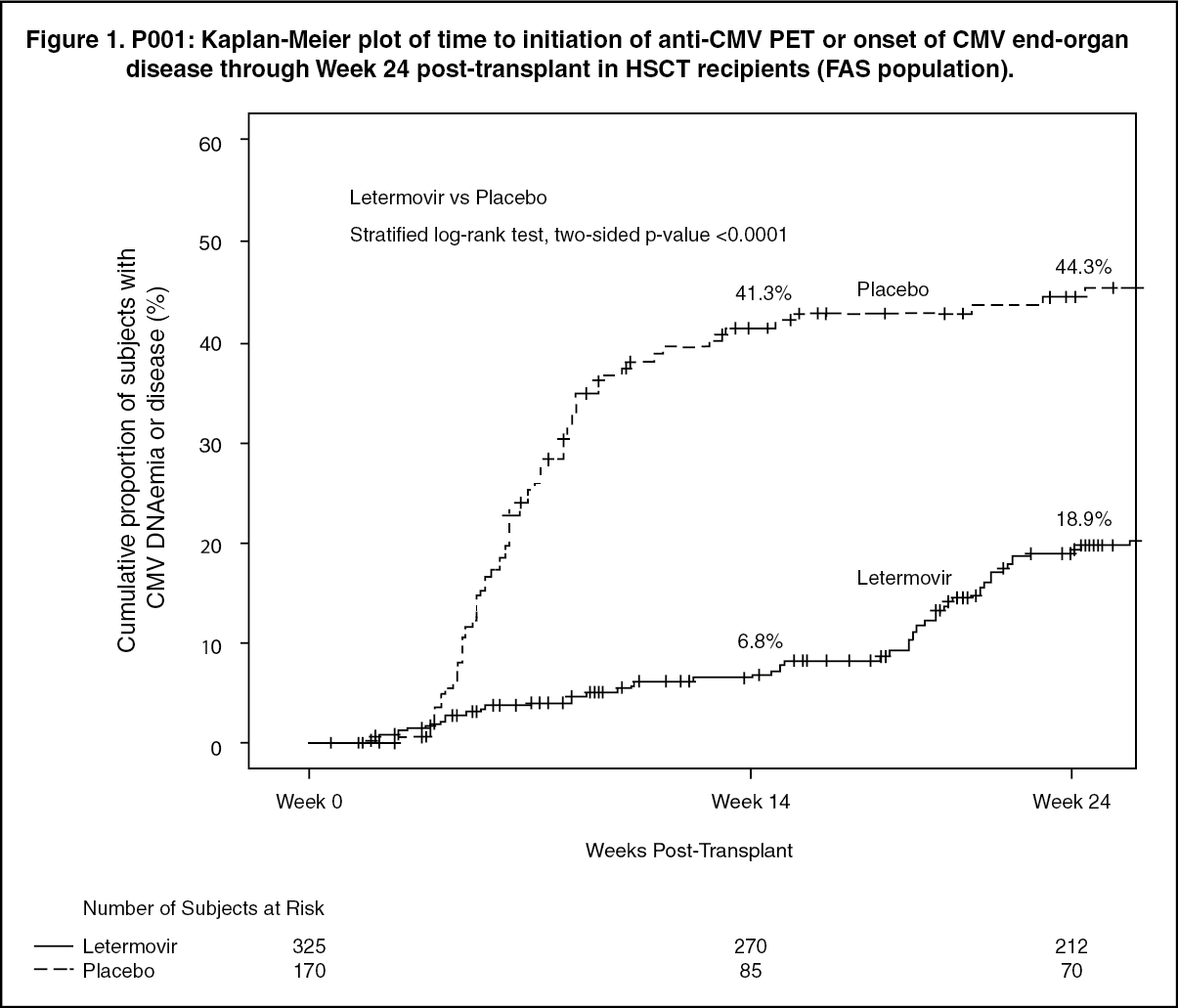 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image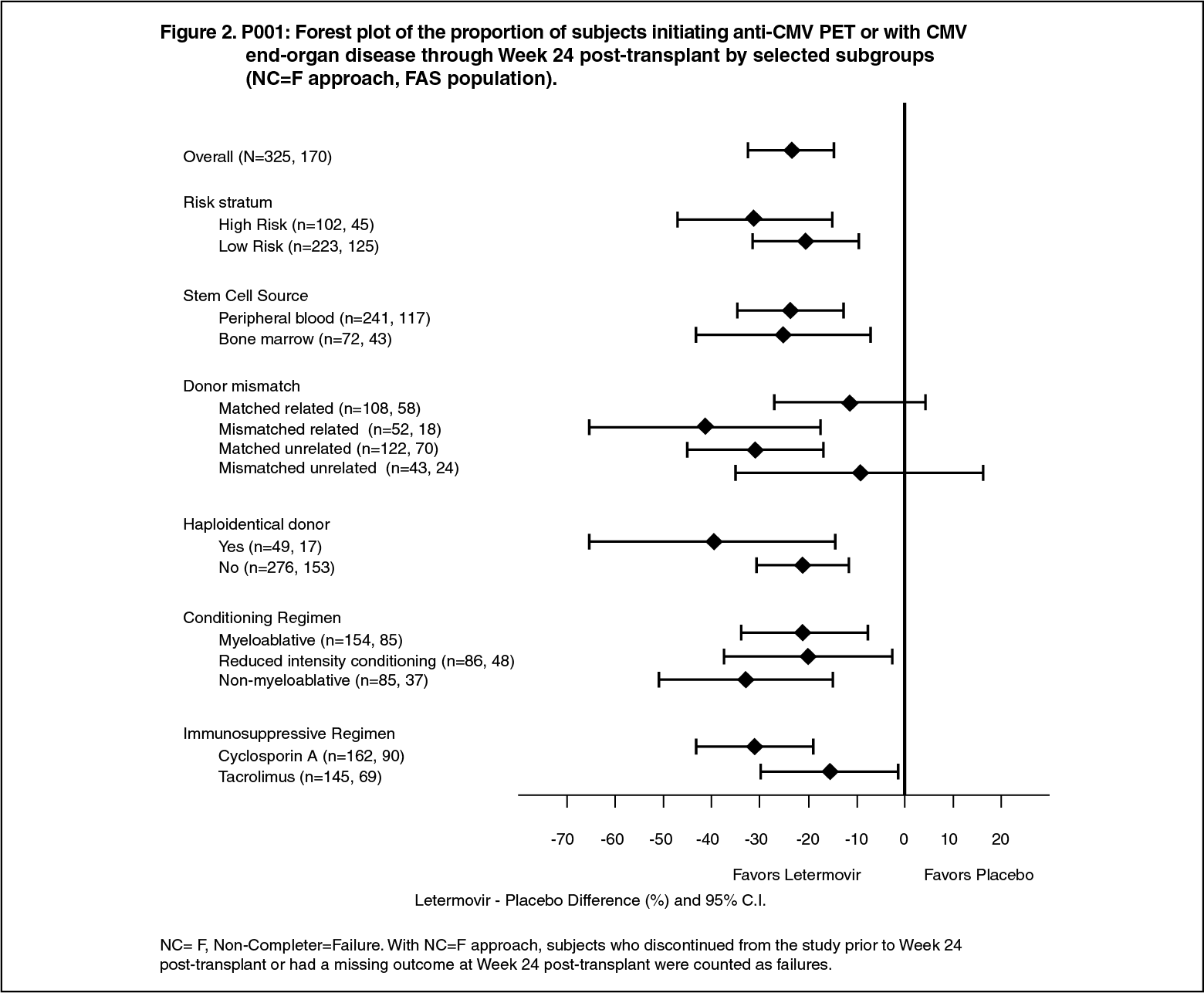 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image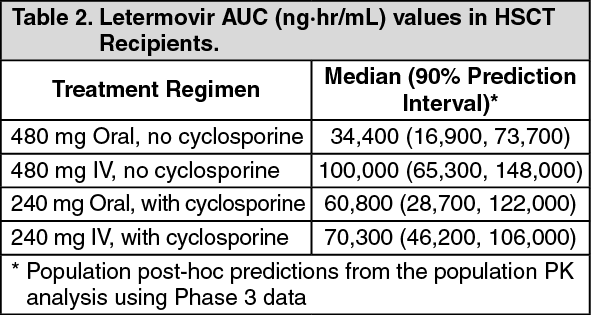 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image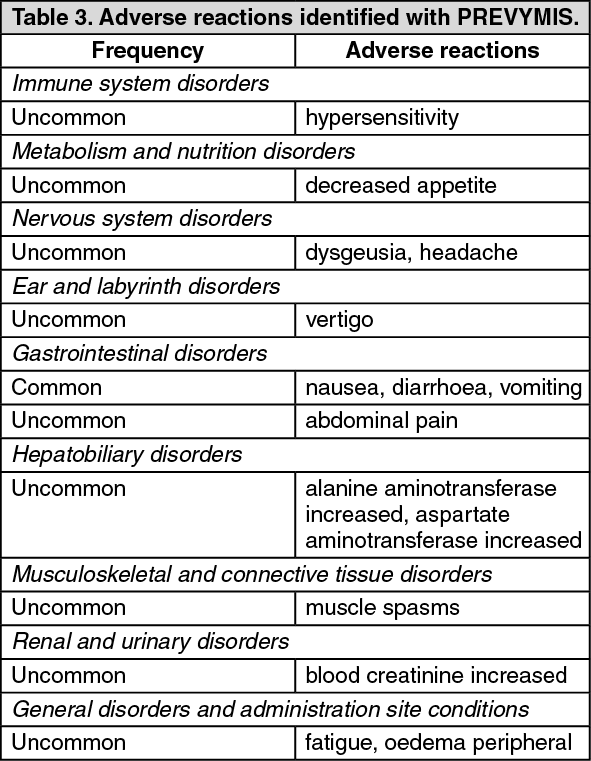 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image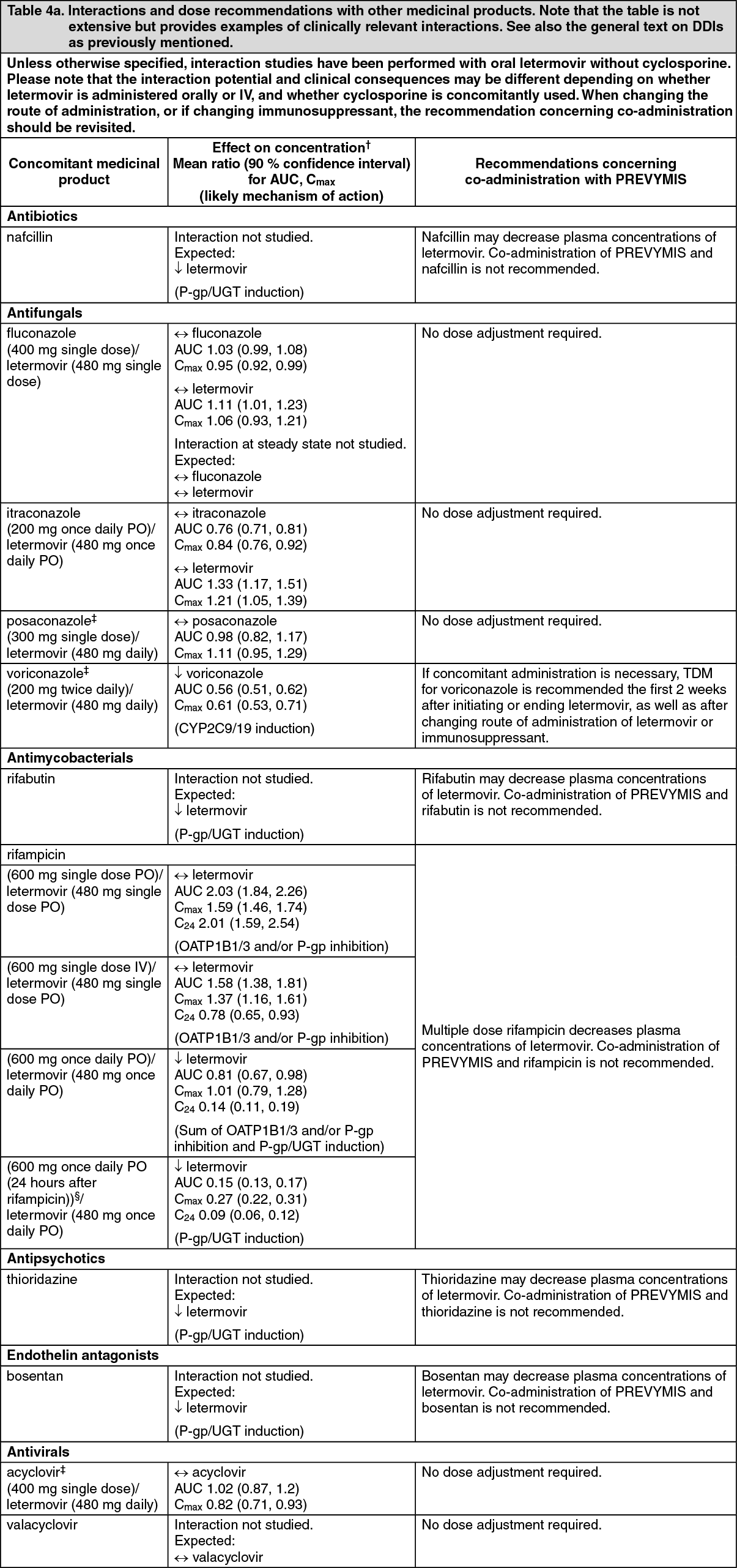 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image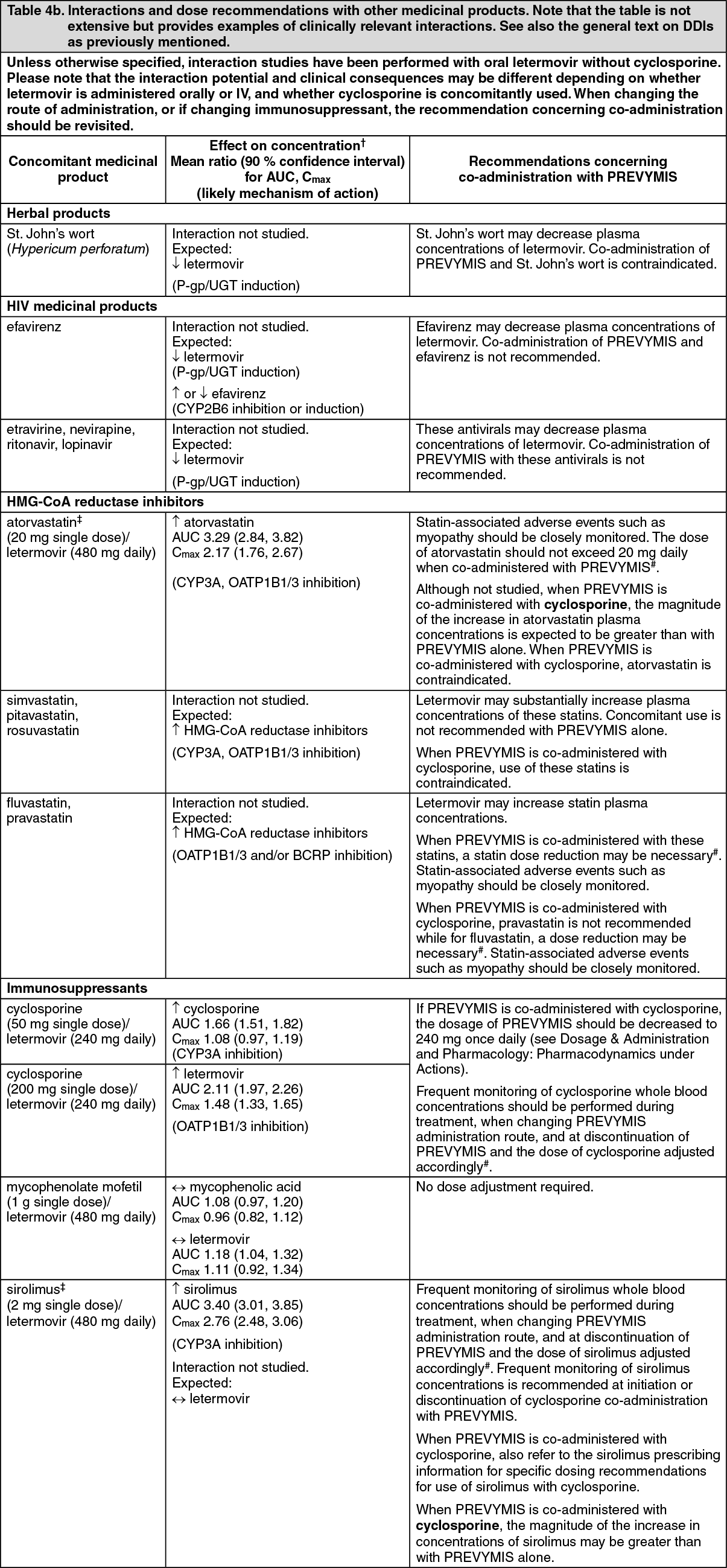 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image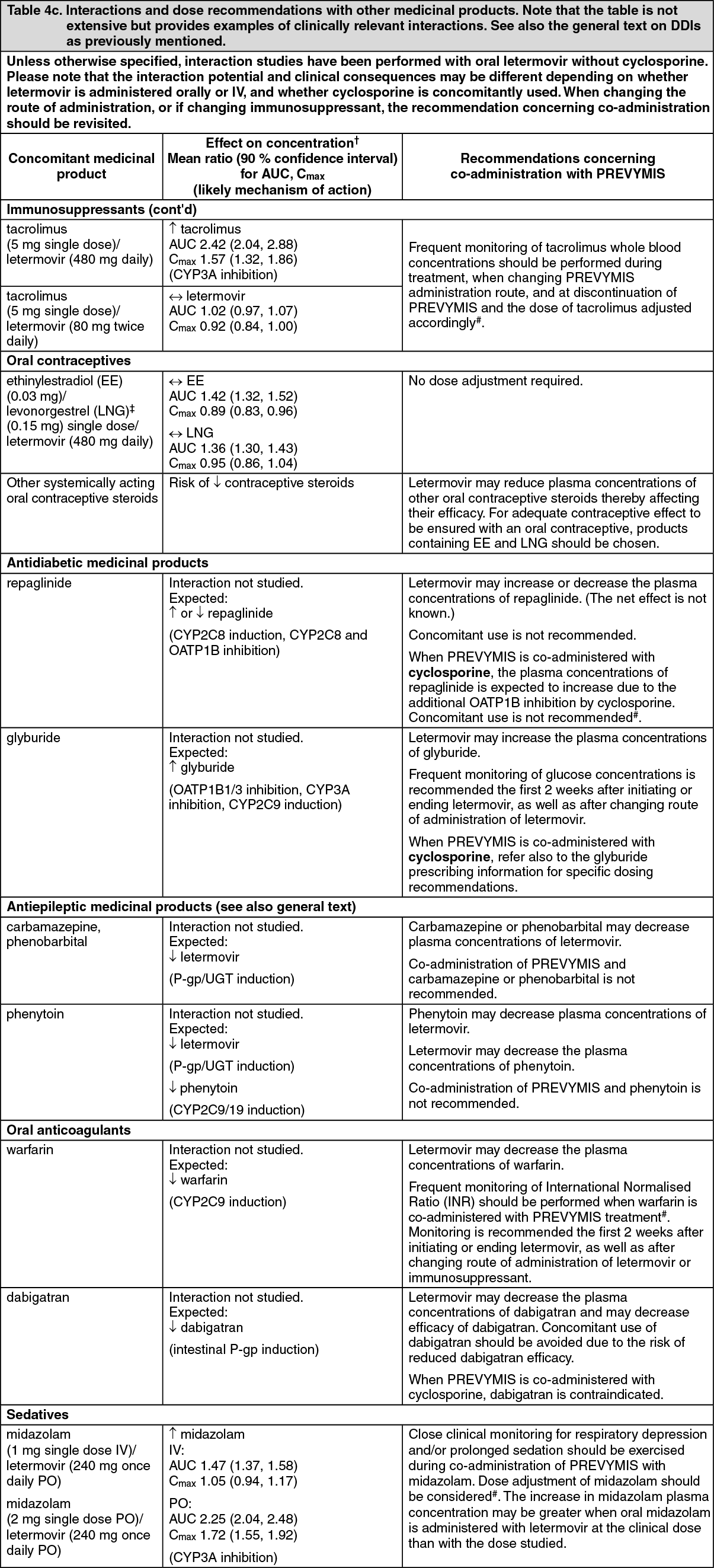 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image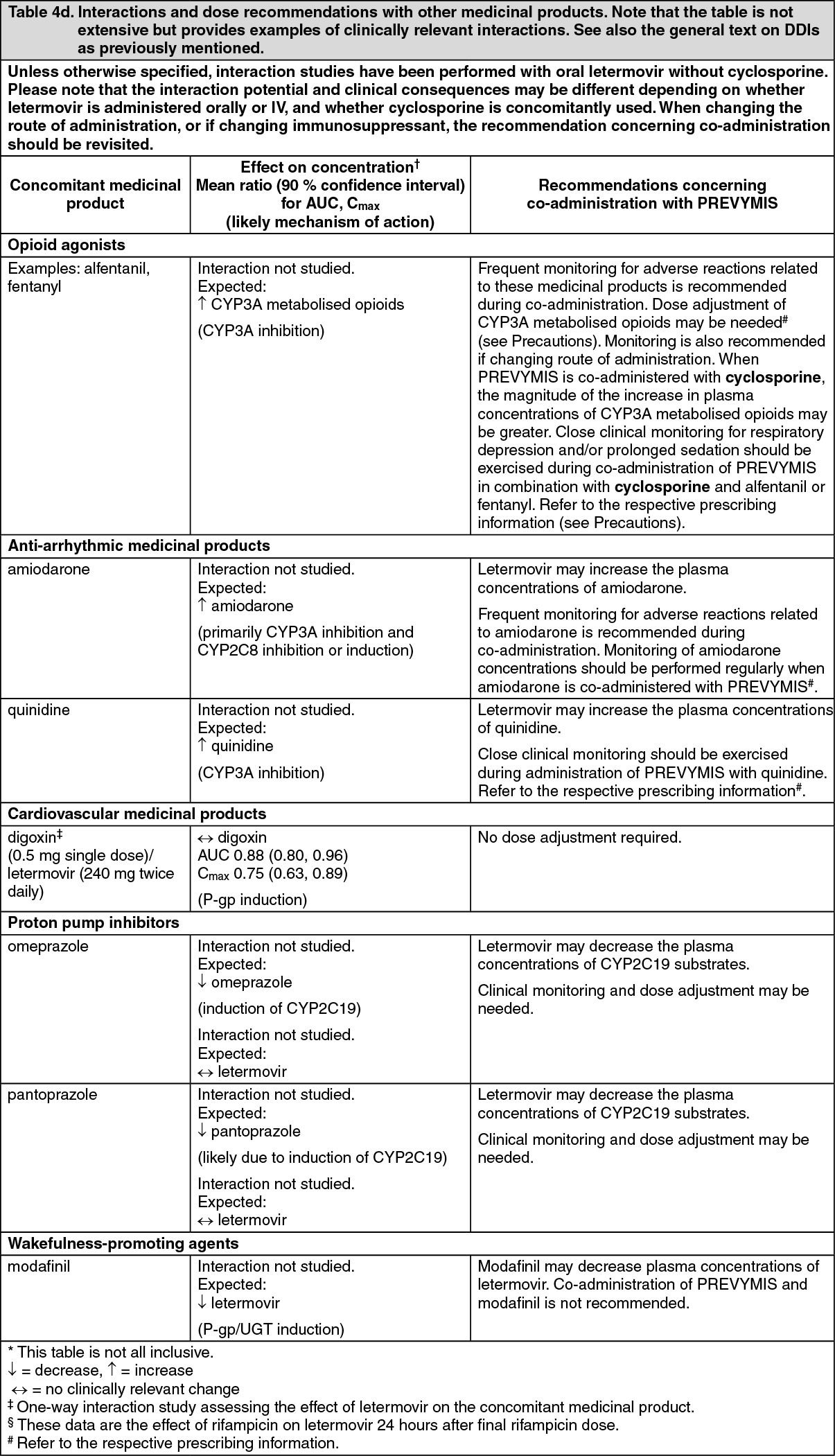 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
