


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Rilpivirine is a diarylpyrimidine non-nucleoside reverse-transcriptase inhibitor (NNRTI) of HIV-1. Rilpivirine activity is mediated by non-competitive inhibition of HIV-1 reverse transcriptase (RT). Rilpivirine does not inhibit the human cellular DNA polymerases α, β and γ.
Antiviral activity in vitro: Rilpivirine exhibited activity against laboratory strains of wild-type HIV-1 in an acutely infected T-cell line with a median EC50 value for HIV-1/IIIB of 0.73 nM (0.27 ng/mL). Although rilpivirine demonstrated limited in vitro activity against HIV-2 with EC50 values ranging from 2,510 to 10,830 nM (920 to 3,970 ng/mL), treatment of HIV-2 infection with rilpivirine is not recommended in the absence of clinical data.
Rilpivirine also demonstrated antiviral activity against a broad panel of HIV-1 group M (subtype A, B, C, D, F, G, H) primary isolates with EC50 values ranging from 0.07 to 1.01 nM (0.03 to 0.37 ng/mL) and group O primary isolates with EC50 values ranging from 2.88 to 8.45 nM (1.06 to 3.10 ng/mL).
Resistance: Considering all of the available in vitro data and in vivo data generated with oral rilpivirine in previously untreated patients, the following resistance-associated mutations, when present at baseline, may affect the activity of rilpivirine: K101E, K101P, E138A, E138G, E138K, E138R, E138Q, V179L, Y181C, Y181I, Y181V, Y188L, H221Y, F227C, M230I, M230L, and the combination of L100I and K103N.
In cell culture: Rilpivirine-resistant strains were selected in cell culture starting from wild-type HIV-1 of different origins and subtypes as well as NNRTI resistant HIV-1. The most commonly observed resistance-associated mutations that emerged included L100I, K101E, V108I, E138K, V179F, Y181C, H221Y, F227C and M230I.
Virologically suppressed patients: The number of subjects who met confirmed virologic failure (CVF) criteria was low across the pooled Phase 3 studies ATLAS and FLAIR. There were 7 CVFs on rilpivirine plus cabotegravir (7/591, 1.2%) and 7 CVFs on current antiretroviral regimen (7/591, 1.2%) through week 48. In the rilpivirine plus cabotegravir group in the pooled analysis, 5/591 (0.8%) subjects had resistance development: 5/591 (0.8%) and 4/591 (0.7%) with resistance-associated mutations to rilpivirine (K101E [n=1], E138A/E/K/T [n=1], E138A [n=1], or E138K [n=2]) and/or cabotegravir (G140R [n=1], Q148R [n=2], or N155H [n=1]), respectively. The 4 CVFs on cabotegravir plus rilpivirine in FLAIR had HIV-1 subtype A1 (n=3) or AG (n=1). One CVF in FLAIR never received an injection. The 3 CVFs on cabotegravir plus rilpivirine in ATLAS had HIV-1 subtype A, A1, or AG. In 2 of these 3 CVFs the rilpivirine resistance-associated mutations observed at failure were also observed at baseline in PBMC HIV-1 DNA.
In the ATLAS-2M study 10 subjects met CVF criteria through week 48: 8/522 (1.5%) in the Q8W arm and 2/523 (0.4%) in the Q4W arm. In the Q8W group 5/522 (1.0%) had resistance development: 4/522 (0.8%) and 5/522 (1.0%) with resistance-associated mutations to rilpivirine (E138A [n=1], E138K [n=1], K101E [n=2], or Y188L [n=1]) and/or cabotegravir (Q148R [n=3] or N155H [n=4]), respectively. In the Q4W group 2/523 (0.4%) had resistance development: 1/523 (0.2%) and 2/523 (0.4%) had rilpivirine (K101E [n=1], M230L [n=1]) and/or cabotegravir (E138K [n=1], Q148R [n=1], or N155H [n=1]) resistance-associated mutations, respectively. At baseline in the Q8W arm, 5 subjects had rilpivirine resistance-associated mutations and 1 of those subjects carried a cabotegravir resistance-associated mutation. Neither subject in the Q4W arm had any rilpivirine or cabotegravir resistance-associated mutation at baseline. The 10 CVFs on cabotegravir plus rilpivirine in ATLAS-2M had HIV-1 subtype A (n=1), A1 (n=2), B (n=4), C (n=2), or Complex (n=1).
Cross-resistance: Site-directed NNRTI mutant virus: In a panel of 67 HIV-1 recombinant laboratory strains with one mutation at RT positions associated with NNRTI resistance, including the most commonly found K103N and Y181C, rilpivirine showed antiviral activity against 64 (96%) of these strains. The single resistance-associated mutations associated with a loss of susceptibility to rilpivirine were: K101P, Y181I and Y181V. The K103N mutation did not result in reduced susceptibility to rilpivirine by itself, but the combination of K103N and L100I resulted in a 7-fold reduced susceptibility to rilpivirine.
Recombinant clinical isolates: Rilpivirine retained sensitivity (fold change ≤ biological cut-off) against 62% of 4,786 HIV-1 recombinant clinical isolates resistant to efavirenz and/or nevirapine.
Virologically suppressed patients: In the week 48 analysis of the Phase 3 studies ATLAS and FLAIR, 5/7 CVFs had phenotypic resistance against rilpivirine at failure. Among these 5 patients, phenotypic cross-resistance was observed against efavirenz (n=4), etravirine (n=3), and nevirapine (n=4).
Effects on electrocardiogram: No effect on QTcF interval was shown for oral rilpivirine at the recommended dose of 25 mg once daily in a randomised, placebo and active (moxifloxacin 400 mg once daily) controlled crossover study in 60 healthy adults, with 13 measurements over 24 hours at steady-state. Plasma rilpivirine concentrations after REKAMBYS injections are comparable to those achieved with oral rilpivirine at dose of 25 mg qd. REKAMBYS at the recommended dose of 600 mg monthly or 900 mg every 2 months is not associated with a clinically relevant effect on QTc.
When supratherapeutic doses of 75 mg once daily and 300 mg once daily of oral rilpivirine were studied in healthy adults, the maximum mean time-matched (95% upper confidence bound) differences in QTcF interval from placebo after baseline correction were 10.7 (15.3) and 23.3 (28.4) ms, respectively. Steady-state administration of oral rilpivirine 75 mg once daily and 300 mg once daily resulted in a mean Cmax approximately 4.4-fold and 11.6-fold, respectively, higher than the mean steady-state Cmax observed with the recommended 600 mg once monthly dose of REKAMBYS. Steady state administration of oral rilpivirine 75 mg once daily and 300 mg once daily resulted in a mean Cmax approximately 4.1-fold and 10.7-fold, respectively, higher than the mean steady state Cmax observed with the recommended 900 mg every 2 months dose of REKAMBYS.
Clinical efficacy and safety: Every 1 month dosing: The efficacy of REKAMBYS plus cabotegravir injection has been evaluated in two Phase 3 randomised, multicentre, active-controlled, parallel-arm, open-label, non-inferiority studies, FLAIR (201584) and ATLAS (201585). The primary analysis was conducted after all subjects completed their week 48 visit or discontinued the study prematurely.
Patients virologically suppressed (on prior dolutegravir-based regimen for 20 weeks): In FLAIR, 629 HIV-1-infected, antiretroviral treatment (ART)-naïve subjects received a dolutegravir integrase strand transfer inhibitor (INI) containing regimen for 20 weeks (either dolutegravir/abacavir/lamivudine or dolutegravir + 2 other nucleoside reverse transcriptase inhibitors if subjects were HLA-B*5701 positive). Subjects who were virologically suppressed (HIV-1 RNA <50 copies per mL, n=566) were then randomised (1:1) to receive either a rilpivirine plus cabotegravir regimen or remain on the CAR. Subjects randomised to receive the rilpivirine plus cabotegravir regimen, initiated treatment with oral lead-in dosing with a cabotegravir (30 mg) tablet plus a rilpivirine (25 mg) tablet once daily for at least 4 weeks, followed by treatment with cabotegravir injection (month 1: 600 mg, month 2 onwards: 400 mg injection) plus rilpivirine injection (month 1: 900 mg injection, month 2 onwards: 600 mg injection), monthly, for up to 96 weeks.
Patients virologically suppressed (stable on prior ART for at least 6 months): In ATLAS, 616 HIV-1-infected, ART-experienced, virologically-suppressed (for at least 6 months) subjects (HIV-1 RNA <50 copies per mL) were randomised (1:1) and received either a rilpivirine plus cabotegravir regimen or remained on the CAR. Subjects randomised to receive the rilpivirine plus cabotegravir regimen initiated treatment with oral lead-in dosing with a cabotegravir (30 mg) tablet plus a rilpivirine (25 mg) tablet once daily for at least 4 weeks, followed by treatment with cabotegravir injection (month 1: 600 mg, month 2 onwards: 400 mg injection) plus rilpivirine injection (month 1: 900 mg injection, month 2 onwards: 600 mg injection), monthly, for an additional 44 weeks. In ATLAS, 50%, 17%, and 33% of subjects received an NNRTI, PI, or INI (respectively) as their baseline third treatment agent class prior to randomisation and this was similar between treatment arms.
Pooled Phase 3 studies: At baseline, in the pooled analysis, in the rilpivirine plus cabotegravir arm the median age of subjects was 38 years, 27% were female, 27% were non-white, 1% were ≥65 years and 7% had CD4+ cell count less than 350 cells per mm3; these characteristics were similar between treatment arms.
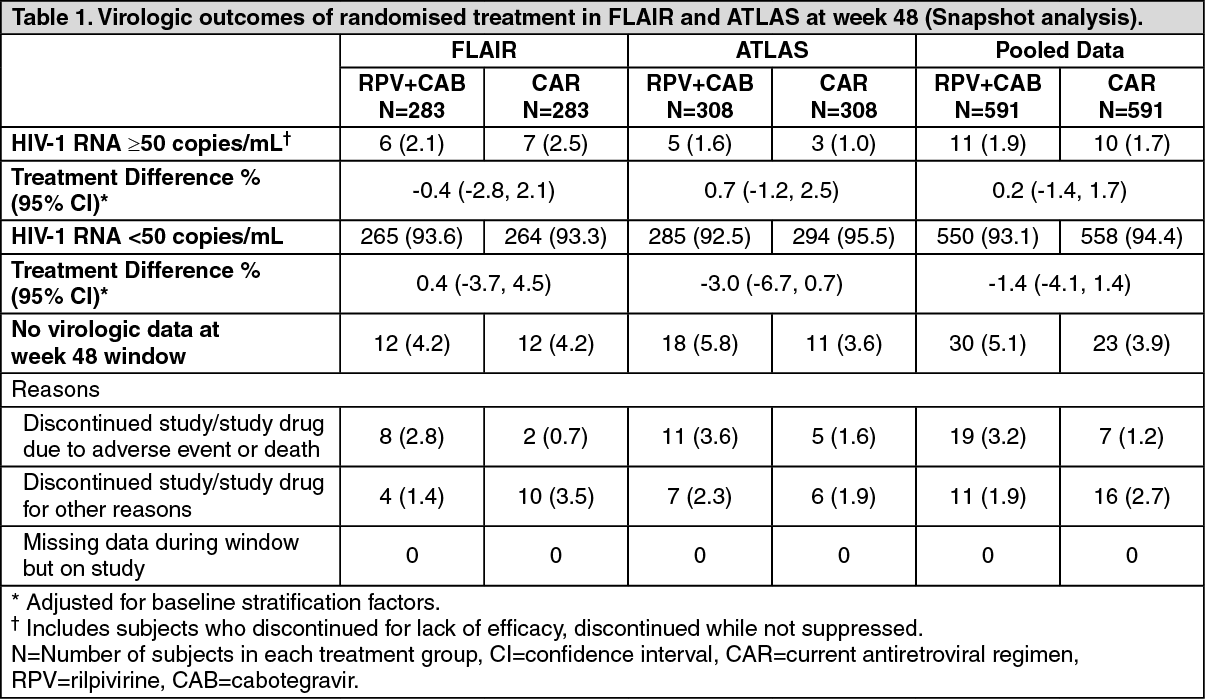
The primary endpoint of both studies was the proportion of subjects with plasma HIV-1 RNA ≥50 copies/mL at week 48 (snapshot algorithm for the ITT-E population).
In a pooled analysis of the two Phase 3 studies, rilpivirine plus cabotegravir was non-inferior to CAR on the proportion of subjects having plasma HIV-1 RNA ≥50 c/mL (1.9% and 1.7% respectively) at week 48. The adjusted treatment difference between rilpivirine plus cabotegravir and CAR (0.2; 95% CI: -1.4, 1.7) met the non-inferiority criterion (upper bound of the 95% CI below 4%) [see Table 1].
The primary endpoint and other week 48 outcomes, including outcomes by key baseline factors, for FLAIR, ATLAS, and pooled data are shown in Table 1 and Table 2. (See Tables 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image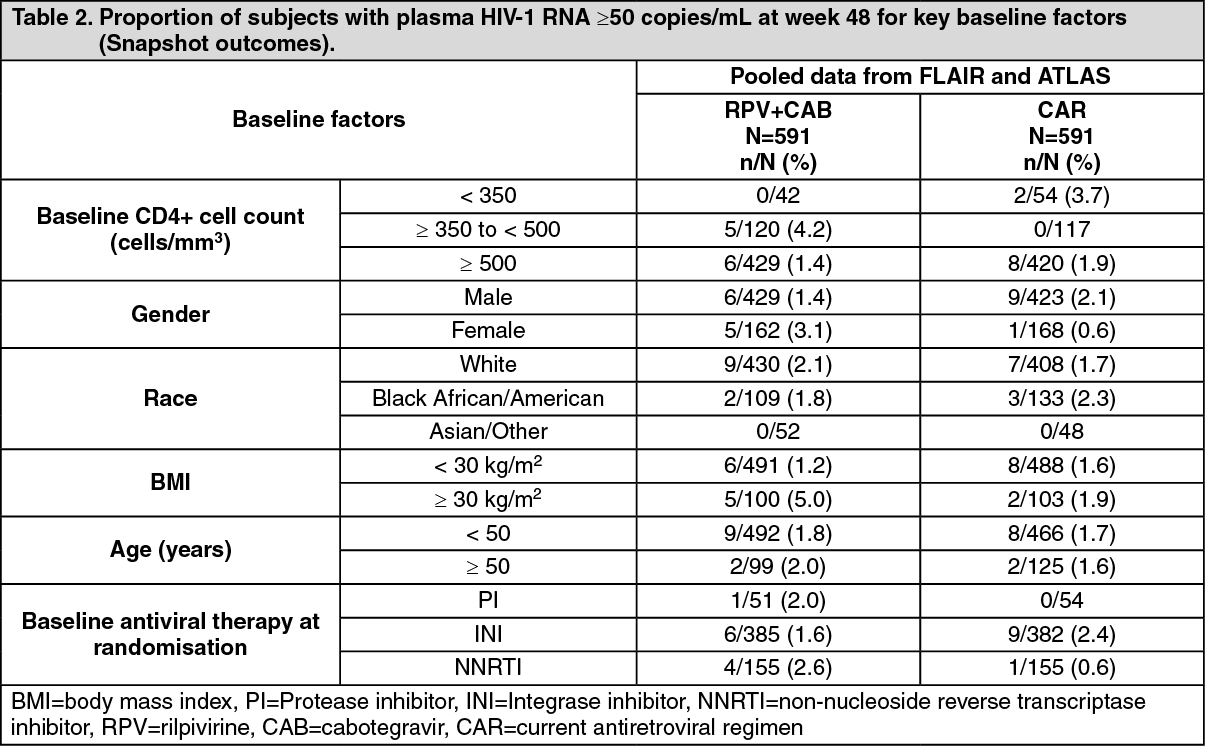 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the FLAIR and ATLAS studies, treatment differences across baseline characteristics (CD4+ count, gender, age, race, BMI, baseline third agent treatment class) were comparable.
In the FLAIR study at 96 Weeks, the results remained consistent with the results at 48 Weeks. The proportion of subjects having plasma HIV-1 RNA ≥50 c/mL in rilpivirine plus cabotegravir (n=283) and CAR (n=283) was 3.2% and 3.2% respectively (adjusted treatment difference between REKAMBYS plus cabotegravir and CAR [0.0; 95% CI: -2.9, 2.9]). The proportion of subjects having plasma HIV-1 RNA <50 c/mL in REKAMBYS plus cabotegravir and CAR was 87% and 89%, respectively (adjusted treatment difference between REKAMBYS plus cabotegravir and CAR [-2.8; 95% CI: -8.2, 2.5]).
Every 2 months dosing: Patients virologically suppressed (stable on prior ART for at least 6 months): The efficacy and safety of rilpivirine injection given every 2 months, has been evaluated in one Phase 3b randomised, multicentre, parallel-arm, open-label, non-inferiority study, ATLAS-2M (207966). The primary analysis was conducted after all subjects completed their week 48 visit or discontinued the study prematurely.
In ATLAS-2M, 1045 HIV-1 infected, ART-experienced, virologically suppressed subjects were randomised (1:1) and received a rilpivirine plus cabotegravir injection regimen administered either every 2 months or monthly. Subjects initially on non-cabotegravir/rilpivirine treatment received oral lead-in treatment comprising one rilpivirine tablet (25 mg) plus one cabotegravir tablet (30 mg), daily, for at least 4 weeks. Subjects randomised to monthly rilpivirine injections (month 1: 900 mg injection, month 2 onwards: 600 mg injection) and cabotegravir injections (month 1: 600 mg injection, month 2 onwards: 400 mg injection administered) received treatment for an additional 44 weeks. Subjects randomised to every 2 months rilpivirine injections (900 mg injection at months 1, 2, 4 and every 2 months thereafter) and cabotegravir injections (600 mg injection at months 1, 2, 4 and every 2 months thereafter) received treatment for an additional 44 weeks. Prior to randomisation, 63%, 13% and 24% of subjects received rilpivirine plus cabotegravir for 0 weeks, 1 to 24 weeks and >24 weeks, respectively.
At baseline, the median age of subjects was 42 years, 27% were female, 27% were non-white, 4% were ≥65 years, and 6% had a CD4+ cell count less than 350 cells per mm3; these characteristics were similar between the treatment arms.
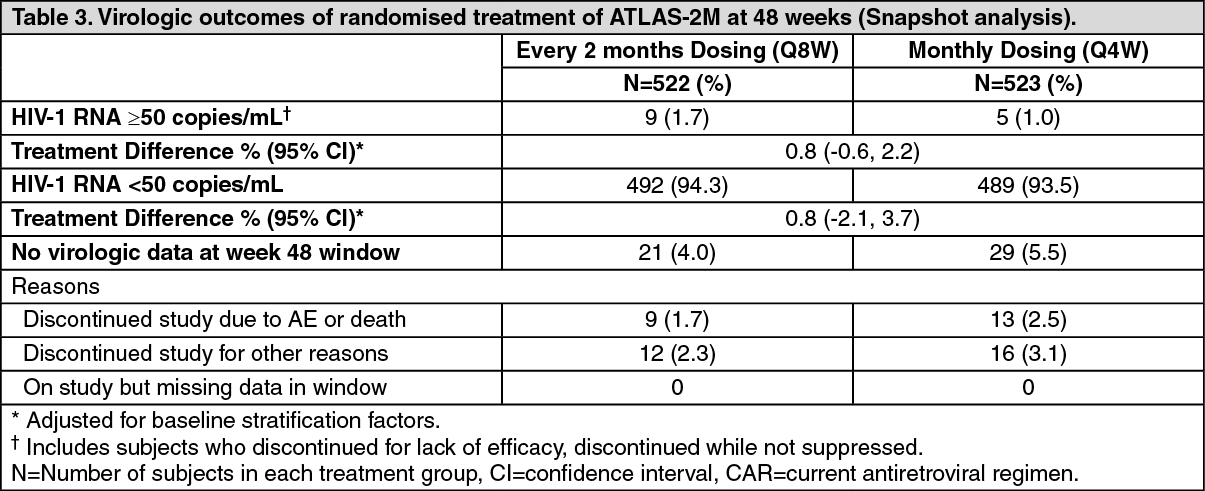
The primary endpoint in ATLAS-2M was the proportion of subjects with a plasma HIV-1 RNA ≥50 c/mL at week 48 (snapshot algorithm for the ITT-E population).
In ATLAS-2M, rilpivirine plus cabotegravir administered every 2 months was non-inferior to cabotegravir and rilpivirine administered every month on the proportion of subjects having plasma HIV-1 RNA ≥50 c/mL (1.7% and 1.0% respectively) at week 48. The adjusted treatment difference between cabotegravir plus rilpivirine administered every 2 months and every month (0.8; 95% CI: -0.6, 2.2) met the non-inferiority criterion (upper bound of the 95% CI below 4%). (See Tables 3 and 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image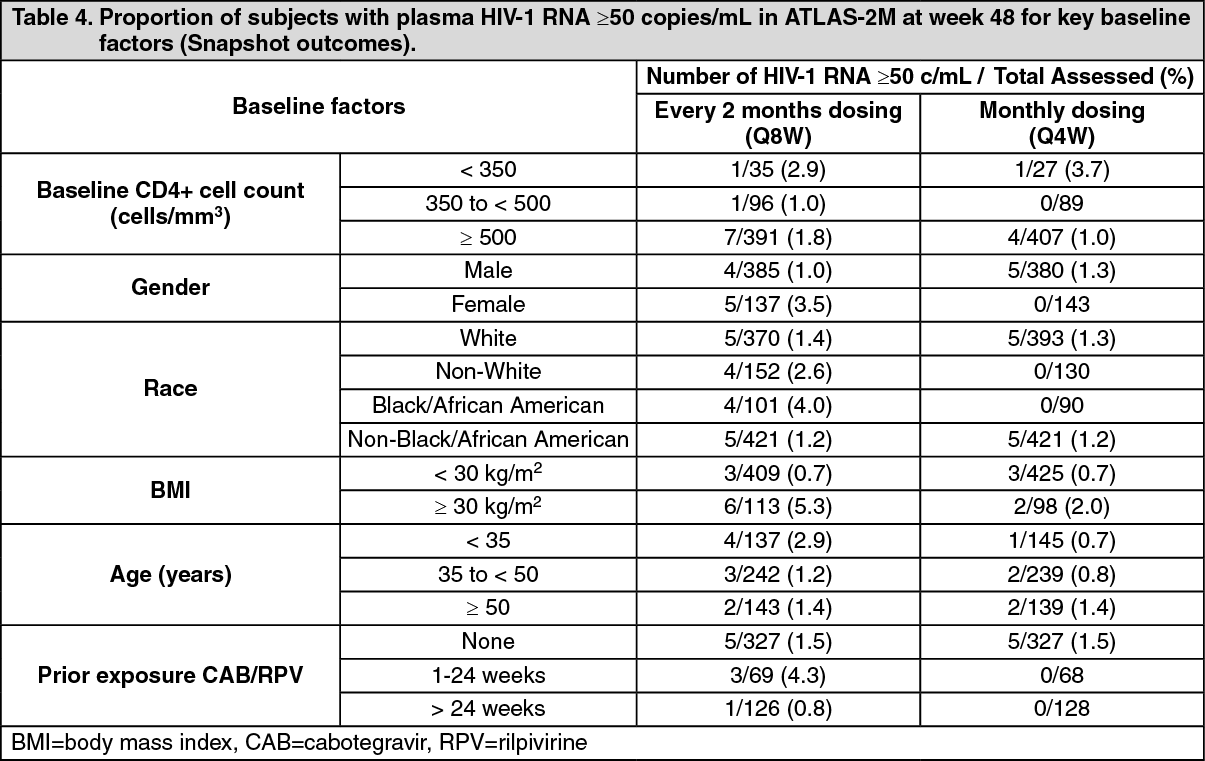 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the ATLAS-2M study, treatment differences on the primary endpoint across baseline characteristics (CD4+ lymphocyte count, gender, race, BMI, age and prior exposure to cabotegravir/rilpivirine) were not clinically meaningful.
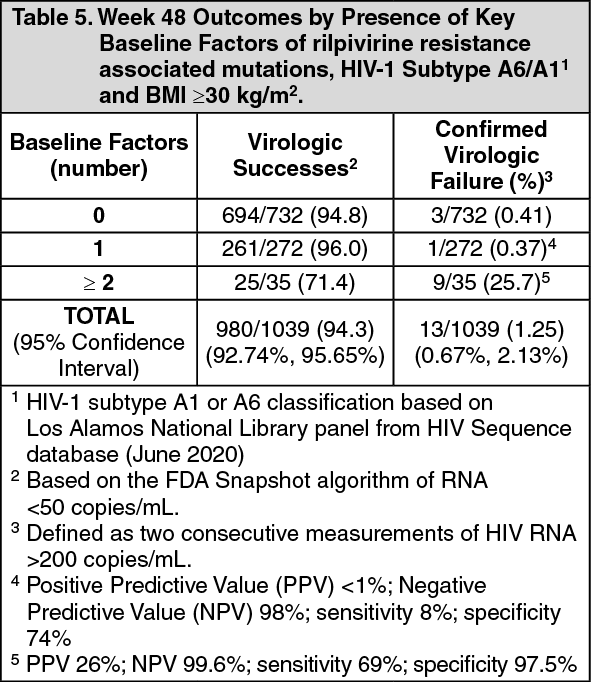
Post-hoc analysis: Multivariable analyses of pooled phase 3 studies (ATLAS, FLAIR, ATLAS-2M), including data from 1039 HIV-infected adults with no prior exposure to rilpivirine plus cabotegravir, examined the influence of the following covariates: baseline viral and participants characteristics, dosing regimen (Q4W or Q8W), and post-baseline plasma drug concentrations on CVF using regression modeling with a covariate selection procedure. Through Week 48 in these studies, 13/1039 (1.25%) participants had CVF while receiving rilpivirine plus cabotegravir.
Four covariates were significantly associated (P < 0.05 for each adjusted odds ratio) with increased risk of CVF: rilpivirine resistance associated mutations (RAMs) at baseline identified by proviral DNA genotypic assay, HIV-1 subtype A6/A1 (associated with integrase L74I polymorphism), rilpivirine trough concentration 4 weeks following initial injection dose, BMI of at least 30 kg/m2 (associated with cabotegravir pharmacokinetics). Other covariates including Q4W or Q8W dosing, female gender, or other viral subtypes (non A6/A1) had no significant association with CVF. No baseline factor, when present in isolation, was predictive of virologic failure. However, a combination of at least 2 of the following baseline factors was associated with increased risk of CVF: rilpivirine resistance associated mutations, HIV-1 subtype A6/A1, or BMI ≥30 kg/m2 (Table 5). (See Table 5.)
 Click on icon to see table/diagram/image
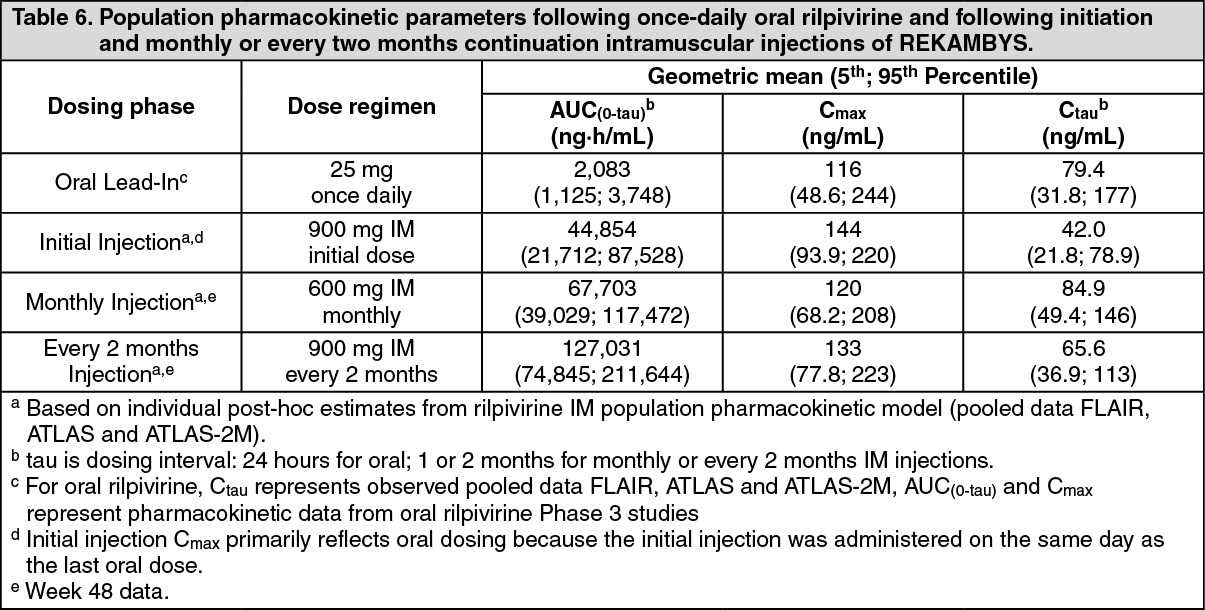
Click on icon to see table/diagram/imagePharmacokinetics: The pharmacokinetic properties of REKAMBYS have been evaluated in healthy and HIV-1 infected adults. (See Table 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAbsorption: Rilpivirine prolonged-release injection exhibits absorption rate-limited kinetics (ie, flip-flop pharmacokinetics) resulting from slow absorption from the gluteal muscle into the systemic circulation resulting in sustained rilpivirine plasma concentrations.
Following a single intramuscular dose, rilpivirine plasma concentrations are detectable the first day and gradually rise to reach maximum plasma concentrations after a median of 3-4 days. Rilpivirine has been detected in plasma up to 52 weeks or longer after administration of a single dose of REKAMBYS. After 1 year of monthly or every 2 months injections, approximately 80% of the rilpivirine pharmacokinetic steady-state exposure is reached.
Plasma rilpivirine exposure increases in proportion or slightly less than in proportion to dose following single and repeat IM injections of doses ranging from 300 to 1200 mg.
Distribution: Rilpivirine is approximately 99.7% bound to plasma proteins in vitro, primarily to albumin. Based on population pharmacokinetics analysis, the typical apparent volume of the central compartment (Vc/F) for rilpivirine after IM administration was estimated to be 132 L, reflecting a moderate distribution to peripheral tissues.
Rilpivirine is present in cerebrospinal fluid (CSF). In HIV-1-infected subjects receiving a regimen of rilpivirine injection plus cabotegravir injection, the median rilpivirine CSF to plasma concentration ratio (n=16) was 1.07 to 1.32% (range: not quantifiable to 1.69%). Consistent with therapeutic rilpivirine concentrations in the CSF, CSF HIV-1 RNA (n=16) was <50 c/mL in 100% and <2 c/mL in 15/16 (94%) of subjects. At the same time point, plasma HIV-1 RNA (n=18) was <50 c/mL in 100% and <2 c/mL in 12/18 (66.7%) of subjects.
Biotransformation: In vitro experiments indicate that rilpivirine primarily undergoes oxidative metabolism mediated by the cytochrome P450 (CYP) 3A system.
Elimination: The mean apparent half-life of rilpivirine following REKAMBYS administration is absorption rate-limited and was estimated to be 13-28 weeks.
The apparent plasma clearance (CL/F) of rilpivirine was estimated to be 5.08 L/h.
After single dose administration of oral 14C-rilpivirine, on average 85% and 6.1% of the radioactivity could be retrieved in faeces and urine, respectively. In faeces, unchanged rilpivirine accounted for on average 25% of the administered dose. Only trace amounts of unchanged rilpivirine (<1% of dose) were detected in urine.
Special patient populations: Gender: No clinically relevant differences in the rilpivirine exposure after intramuscular (IM) administration have been observed between men and women.
Race: No clinically relevant effect of race on the rilpivirine exposure after intramuscular administration has been observed.
BMI: No clinically relevant effect of BMI on the rilpivirine exposure after intramuscular administration has been observed.
Elderly: No clinically relevant effect of age on the rilpivirine exposure after intramuscular administration has been observed. Pharmacokinetic data for rilpivirine in subjects of >65 years old are limited.
Renal impairment: The pharmacokinetics of rilpivirine have not been studied in patients with renal insufficiency. Renal elimination of rilpivirine is negligible. No dose adjustment is needed for patients with mild or moderate renal impairment. In patients with severe renal impairment or end-stage renal disease, REKAMBYS should be used with caution, as plasma concentrations may be increased due to alteration of drug absorption, distribution and/or metabolism secondary to renal dysfunction. In patients with severe renal impairment or end-stage renal disease, the combination of REKAMBYS with a strong CYP3A inhibitor should only be used if the benefit outweighs the risk. As rilpivirine is highly bound to plasma proteins, it is unlikely that it will be significantly removed by haemodialysis or peritoneal dialysis (see Dosage & Administration).
Hepatic impairment: Rilpivirine is primarily metabolised and eliminated by the liver. In a study comparing 8 patients with mild hepatic impairment (Child-Pugh score A) to 8 matched controls, and 8 patients with moderate hepatic impairment (Child-Pugh score B) to 8 matched controls, the multiple dose exposure of oral rilpivirine was 47% higher in patients with mild hepatic impairment and 5% higher in patients with moderate hepatic impairment. However, it may not be excluded that the pharmacologically active, unbound, rilpivirine exposure is significantly increased in moderate hepatic impairment. No dose adjustment is suggested but caution is advised in patients with moderate hepatic impairment. REKAMBYS has not been studied in patients with severe hepatic impairment (Child-Pugh score C). Therefore, REKAMBYS is not recommended in patients with severe hepatic impairment (see Dosage & Administration).
HBV/HCV Co-infected Patients: Population pharmacokinetic analysis indicated that hepatitis B and/or C virus co-infection had no clinically relevant effect on the rilpivirine exposure after oral rilpivirine intake.
Paediatric Patients: The phamacokinetics of rilpivirine in children and adolescents aged <18 years have not been established with REKAMBYS.
Toxicology: Preclinical safety data: All studies were performed with rilpivirine for oral use except for the studies on local tolerance with REKAMBYS injections.
Repeated dose toxicity: Liver toxicity associated with liver enzyme induction was observed in rodents. In dogs, cholestasis-like effects were noted.
Reproductive toxicology studies: Studies in animals have shown no evidence of relevant embryonic or foetal toxicity or an effect on reproductive function. There was no teratogenicity with oral rilpivirine in rats and rabbits. The exposures at the embryo-foetal No Observed Adverse Effects Levels (NOAELs) in rats and rabbits were respectively ≥12 times and ≥57 times the exposure in humans at the maximum recommended human daily dose of 25 mg once daily in HIV-1 infected patients or 600 mg or 900 mg intramuscular injection dose of rilpivirine long-acting injectable suspension.
Carcinogenesis and mutagenesis: Oral rilpivirine was evaluated for carcinogenic potential by oral gavage administration to mice and rats up to 104 weeks. At the lowest tested doses in the carcinogenicity studies, the systemic exposures (based on AUC) to rilpivirine were ≥17 times (mice) and ≥2 times (rats) the exposure in humans at the maximum recommended human daily dose of 25 mg once daily in HIV-1 infected patients or 600 mg or 900 mg intramuscular injection dose of rilpivirine long-acting injectable suspension. In rats, there were no drug-related neoplasms. In mice, rilpivirine was positive for hepatocellular neoplasms in both males and females. The observed hepatocellular findings in mice may be rodent-specific.
Rilpivirine has tested negative in the absence and presence of a metabolic activation system in the in vitro Ames reverse mutation assay and the in vitro clastogenicity mouse lymphoma assay. Rilpivirine did not induce chromosomal damage in the in vivo micronucleus test in mice.
Local tolerance for REKAMBYS: After long-term repeated IM administration of REKAMBYS in dogs and minipigs, slight, short-lasting (ie, 1-4 days in minipigs) erythema was observed, and white deposits were noted at the injection sites at necropsy, accompanied by swelling and discoloration of draining lymph nodes. Microscopic examination showed macrophage infiltration and eosinophilic deposits at the injection sites. A macrophage infiltration response was also noted in the draining/regional lymph nodes. These findings were considered to be a reaction to the deposited material rather than a manifestation of local irritation.