


 Sign Out
Sign Out
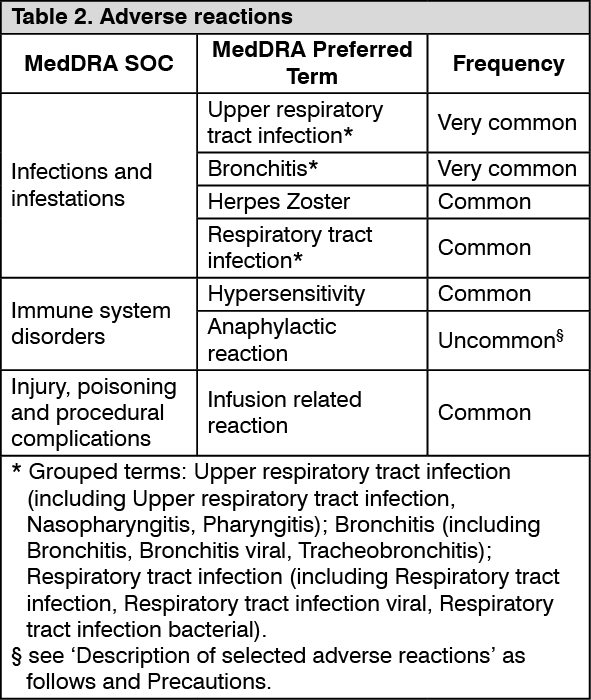
Tabulated list of adverse reactions: Adverse reactions reported from controlled clinical trials are classified by MedDRA System Organ Class (SOC), see Table 2. Within each SOC, preferred terms are arranged by decreasing frequency and then by decreasing seriousness. Frequencies of occurrence of adverse reactions are defined as: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1 000 to <1/100); rare (≥1/10 000 to <1/1 000); very rare (<1/10 000) and not known (cannot be estimated from available data). (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Hypersensitivity and infusion-related reactions: The incidence of hypersensitivity reactions was 2.8% in the anifrolumab group and 0.6% in the placebo group. All hypersensitivity reactions were reported within the first 6 infusions. Hypersensitivity reactions were predominantly mild to moderate in intensity and did not lead to discontinuation of anifrolumab therapy. One serious adverse reaction of hypersensitivity was reported during the patient's first infusion; the patient continued to receive anifrolumab with premedication given for subsequent infusions.
In the SLE development program, anaphylactic reaction was reported for 0.1% (1/837) of patients; the event occurred following the administration of 150 mg anifrolumab, the patient was treated and recovered (see Precautions).
The incidence of infusion-related reactions was 9.4% in the anifrolumab group and 7.1% in the placebo group. Infusion-related reactions were mild or moderate in intensity (the most common symptoms were headache, nausea, vomiting, fatigue, and dizziness); none were serious, and none led to discontinuation of anifrolumab. Infusion-related reactions were most commonly reported at the start of treatment, on the first and second infusions, with fewer reports on subsequent infusions.
Respiratory infections: Reporting rates for anifrolumab compared to placebo were: upper respiratory tract infection (34.4% vs 23.2%), bronchitis (10.7% vs 5.2%) and respiratory tract infection (3.3% vs 1.5%). Infections were predominantly non-serious, mild or moderate in intensity and resolved without discontinuation of anifrolumab therapy (see Precautions).
Herpes zoster: The incidence of herpes zoster infections was 6.1% in the anifrolumab group and 1.3% in the placebo group (see Precautions). In the 52 week clinical trials the mean time to onset was 139 days (range 2 - 351 days).
Herpes zoster infections were predominantly of localised cutaneous presentation, mild or moderate in intensity and resolved without discontinuation of anifrolumab therapy. Cases with multidermatomal involvement and cases of disseminated disease (including central nervous system involvement) have been reported (see Precautions).
Immunogenicity: In the Phase III trials, treatment-emergent anti-drug antibodies were detected in 6 out of 352 (1.7%) patients treated with anifrolumab at the recommended dosing regimen during the 60 week study period. Due to methodological limitations, the clinical relevance of this finding is not known.
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are recommended to report any suspected adverse reactions to AstraZeneca.
View ADR Monitoring Form