


 Sign Out
Sign Out
Pharmacology: Spiriva Respimat: Tiotropium is a long-acting, specific antimuscarinic (anticholinergic) agent. It has similar affinity to the muscarinic receptor subtypes M1 to M5 (KD 5-41 pM). In the airways, inhibition by tiotropium of M3-receptors at the smooth muscle results in relaxation. The competitive and reversible nature of antagonism was shown with human and animal origin receptors. In non-clinical in vitro as well as in vivo studies, bronchoprotective effects were dose-dependent. Bronchoprotective effects lasting at least 24 hours were observed in some of the in vivo studies. The long duration of effect of tiotropium is likely to be due to its slow dissociation from M3-receptors. Tiotropium exhibited a significantly longer dissociation half-life from M3 receptors than ipratropium.
Tiotropium, a N-quaternary anticholinergic agent, is topically (broncho-) selective when administered by inhalation. The high potency (IC50 approximately 0.4 nM for M3) and slow receptor dissociation is associated with a significant and long-acting bronchodilation in patients with chronic obstructive pulmonary disease (COPD) and asthma.
The bronchodilation following inhalation of tiotropium is primarily a local effect on the airways, not a systemic one.
Pharmacodynamics: Spiriva: Mechanism of action: Tiotropium bromide is a long-acting, specific, muscarinic receptor antagonist, in clinical medicine often called an anticholinergic. By binding to the muscarinic receptors in the bronchial smooth musculature, tiotropium bromide inhibits the cholinergic (bronchoconstrictive) effects of acetylcholine, released from parasympathetic nerve endings. It has similar affinity to the subtypes of muscarinic receptors, M1 to M5. In the airways, tiotropium bromide competitively and reversibly antagonises the M3 receptors, resulting in relaxation. The effect was dose dependent and lasted longer than 24h. The long duration is probably due to the very slow dissociation from the M3 receptor, exhibiting a significantly longer dissociation half-life than ipratropium. As an N-quaternary anticholinergic, tiotropium bromide is topically (broncho-) selective when administered by inhalation, demonstrating an acceptable therapeutic range before systemic anticholinergic effects may occur.
Pharmacodynamic effects: The bronchodilation is primarily a local effect (on the airways), not a systemic one. Dissociation from M2-receptors is faster than from M3, which in functional in vitro studies, elicited (kinetically controlled) receptor subtype selectivity of M3 over M2. The high potency and slow receptor dissociation found its clinical correlate in significant and long-acting bronchodilation in patients with COPD.
Cardiac electrophysiology: Electrophysiology: In a dedicated QT study involving 53 healthy volunteers, SPIRIVA 18 mcg and 54 mcg (i.e. three times the therapeutic dose) over 12 days did not significantly prolong QT intervals of the ECG.
Clinical efficacy and safety: The clinical development programme included four one-year and two six-month randomised, double-blind studies in 2,663 patients (1,308 receiving tiotropium bromide). The one-year programme consisted of two placebo-controlled trials and two trials with an active control (ipratropium). The two six-month trials were both, salmeterol and placebo controlled. These studies included lung function and health outcome measures of dyspnoea, exacerbations and health-related quality of life.
Lung function: Tiotropium bromide, administered once daily, provided significant improvement in lung function (forced expiratory volume in one second, FEV1 and forced vital capacity, FVC) within 30 minutes following the first dose which was maintained for 24 hours. Pharmacodynamic steady state was reached within one week with the majority of bronchodilation observed by the third day. Tiotropium bromide significantly improved morning and evening PEFR (peak expiratory flow rate) as measured by patient's daily recordings. The bronchodilator effects of tiotropium bromide were maintained throughout the one-year period of administration with no evidence of tolerance.
A randomised, placebo-controlled clinical study in 105 COPD patients demonstrated that bronchodilation was maintained throughout the 24 hour dosing interval in comparison to placebo regardless of whether the drug was administered in the morning or in the evening.
Long-term clinical trials (6 months and 1 year): Dyspnoea, Exercise tolerance: Tiotropium bromide significantly improved dyspnoea (as evaluated using the Transition Dyspnoea Index.). This improvement was maintained throughout the treatment period.
The impact of improvements in dyspnoea on exercise tolerance was investigated in two randomised, double-blind, placebo-controlled trials in 433 patients with moderate to severe COPD. In these trials, six weeks of treatment with SPIRIVA significantly improved symptom-limited exercise endurance time during cycle ergometry at 75% of maximal work capacity by 19.7% (Trial A) and 28.3% (Trial B) compared with placebo.
Health-related Quality of Life: In a 9-month, randomized, double-blind, placebo-controlled clinical trial of 492 patients, SPIRIVA improved health-related quality of life as determined by the St. George's Respiratory Questionnaire (SGRQ) total score. The proportion of patients treated with SPIRIVA which achieved a meaningful improvement in the SGRQ total score (i.e. > 4 units) was 10.9% higher compared with placebo (59.1% in the SPIRIVA groups vs. 48.2% in the placebo group (p=0.029). The mean difference between the groups was 4.19 units (p=0.001; confidence interval: 1.69-6.68). The improvements of the subdomains of the SGRQ-score were 8.19 units for "symptoms", 3.91 units for "activity" and 3.61 units for "impact on daily life". The improvements of all of these separate subdomains were statistically significant.
COPD Exacerbations: In a randomized, double-blind, placebo controlled trial of 1,829 patients with moderate to very severe COPD, tiotropium bromide statistically significantly reduced the proportion of patients who experienced exacerbations of COPD (32.2% to 27.8%) and statistically significantly reduced the number of exacerbations by 19% (1.05 to 0.85 events per patient year of exposure). In addition, 7.0% of patients in the tiotropium bromide group and 9.5% of patients in the placebo group were hospitalized due to a COPD exacerbation (p=0.056). The number of hospitalizations due to COPD was reduced by 30% (0.25 to 0.18 events per patient year of exposure).
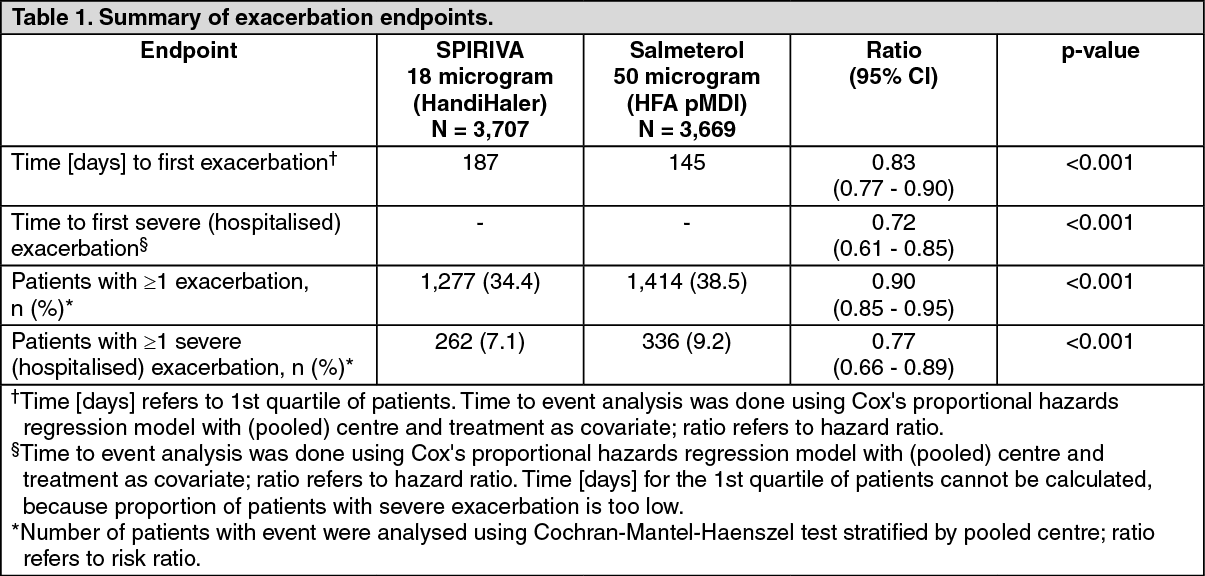
A one-year randomised, double-blind, double-dummy, parallel-group trial compared the effect of treatment with 18 microgram of SPIRIVA once daily with that of 50 microgram of salmeterol HFA pMDI twice daily on the incidence of moderate and severe exacerbations in 7,376 patients with COPD and a history of exacerbations in the preceding year. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageCompared with salmeterol, SPIRIVA increased the time to the first exacerbation (187 days vs. 145 days), with a 17% reduction in risk (hazard ratio, 0.83; 95% confidence interval [CI], 0.77 to 0.90; P<0.001). SPIRIVA also increased the time to the first severe (hospitalised) exacerbation (hazard ratio, 0.72; 95% CI, 0.61 to 0.85; P<0.001).
Long-term clinical trials (more than 1 year, up to 4 years): In a 4-year, randomised, double-blind, placebo-controlled clinical trial of 5,993 randomised patients (3,006 receiving placebo and 2,987 receiving SPIRIVA), the improvement in FEV1 resulting from SPIRIVA, compared with placebo, remained constant throughout 4 years. A higher proportion of patients completed ≥ 45 months of treatment in the SPIRIVA group compared with the placebo group (63.8% vs. 55.4%, p<0.001). The annualized rate of decline of FEV1 compared to placebo was similar between SPIRIVA and placebo. During treatment, there was a 16% reduction in the risk of death. The incidence rate of death was 4.79 per 100 patient years in the placebo group vs. 4.10 per 100 patient years in the tiotropium group (hazard ratio (tiotropium/placebo) = 0.84, 95% CI = 0.73, 0.97). Treatment with tiotropium reduced the risk of respiratory failure (as recorded through adverse event reporting) by 19% (2.09 vs. 1.68 cases per 100 patient years, relative risk (tiotropium/placebo) = 0.81, 95% CI = 0.65, 0.999).
Tiotropium active-controlled study: A long-term, large scale randomised, double-blind, active-controlled study with an observation period up to 3 years has been performed to compare the efficacy and safety of SPIRIVA HandiHaler and SPIRIVA RESPIMAT (5,694 patients receiving SPIRIVA HandiHaler; 5,711 patients receiving SPIRIVA RESPIMAT). The primary endpoints were time to first COPD exacerbation, time to all-cause mortality and in a sub-study (906 patients) trough FEV1 (pre-dose).
The time to first COPD exacerbation was numerically similar during the study with SPIRIVA HandiHaler and SPIRIVA RESPIMAT (hazard ratio (SPIRIVA HandiHaler/SPIRIVA RESPIMAT) 1.02 with a 95% CI of 0.97 to 1.08). The median number of days to the first COPD exacerbation was 719 days for SPIRIVA HandiHaler and 756 days for SPIRIVA RESPIMAT.
The bronchodilator effect of SPIRIVA HandiHaler was sustained over 120 weeks, and was similar to SPIRIVA RESPIMAT. The mean difference in trough FEV1 for SPIRIVA HandiHaler versus SPIRIVA RESPIMAT was 0.010 L (95% CI -0.018 to 0.038 L).
In the post-marketing TIOSPIR study comparing SPIRIVA RESPIMAT and SPIRIVA HandiHaler, all-cause mortality including vital status follow up was similar during the study with SPIRIVA HandiHaler and SPIRIVA RESPIMAT (hazard ratio (SPIRIVA HandiHaler/SPIRIVA RESPIMAT) 1.04 with a 95% CI of 0.91 to 1.19).
Pediatric population: No data in paediatric population were established (see Dosage & Administration).
Spiriva Respimat: Clinical Trials: COPD: The clinical Phase III programme for COPD included two 1-year, two 12-week and two 4-week randomised, double-blind studies in 2901 patients with COPD (1038 receiving the 5 micrograms tiotropium dose). The 1-year programme consisted of two placebo-controlled trials. The two 12-week trials were both active (ipratropium)- and placebo-controlled. All six studies included lung function measurements, with trough FEV1 (i.e. FEV1 measured approximately 10 minutes before the final dose) as the primary endpoint. In addition, the two 1-year studies included health outcome measures of health-related quality of life, dyspnoea, and effect on exacerbations as co-primary endpoints.
Placebo-controlled studies: Lung function: SPIRIVA RESPIMAT administered once daily, provided significant improvement in lung function (forced expiratory volume in one second and forced vital capacity) within 30 minutes following the first dose, compared to placebo. Improvement of lung function was maintained for 24 hours at steady state. Pharmacodynamic steady state was reached within one week.
Mean trough FEV1 treatment difference for SPIRIVA RESPIMAT over placebo in the combined 1-year trials at day 337 was 127 mL (p<0.0001 vs. placebo). Improvement of lung function was maintained for 24 hours at steady state. Pharmacodynamic steady state was reached within one week. The bronchodilator effects of SPIRIVA RESPIMAT were maintained throughout the 48-week period of administration with no evidence of tolerance.
Mean trough FEV1 treatment differences for the combined 12-week trials at day 85 was 118 mL for SPIRIVA RESPIMAT over placebo (p<0.0001) and 64 mL for SPIRIVA RESPIMAT over ipratropium bromide (p=0.0060).
A combined analysis of two randomised, placebo-controlled, crossover, clinical studies demonstrated that the bronchodilator response as measured by mean trough FEV1 for SPIRIVA RESPIMAT was 29 mL higher than Spiriva HandiHaler (18 micrograms) inhalation powder after a 4-week treatment period (p=0.03). Since steady state efficacy is reached within 4 weeks, no longer term study comparing the two products has been conducted.
SPIRIVA RESPIMAT significantly improved morning and evening PEFR (peak expiratory flow rate) as measured by patient's daily recordings (morning improvement mean 22 L/min, p<0.0001; evening improvement mean 26 L/min, p<0.0001). The use of SPIRIVA RESPIMAT resulted in a reduction of rescue bronchodilator use compared to placebo.
Dyspnoea, Health-related Quality of Life, COPD Exacerbations in long-term 1 year studies: (a) SPIRIVA RESPIMAT significantly improved dyspnoea (as evaluated using the Transition Dyspnoea Index) the magnitude of change being 1.05 units at day 337 (p<0.0001 vs. placebo). The mean Baseline Dyspnoea Index was 6.41 units. Improvement was maintained throughout the treatment period.
(b) Patients' evaluation of their Quality of Life (as measured using the St. George's Respiratory Questionnaire) showed that SPIRIVA RESPIMAT had positive effects on the psychosocial impacts of COPD, activities affected by COPD and distress due to COPD symptoms.
The improvement in mean total score between SPIRIVA RESPIMAT versus placebo at the end of the two 1-year studies was statistically significant and maintained throughout the treatment period. By day 337 the mean treatment difference improvement in SGRQ total score from placebo (pooled data from the two 1-year studies) was 3.5 for SPIRIVA RESPIMAT (p<0.0001 vs. placebo). The mean SGRQ total score at baseline was 44.8.
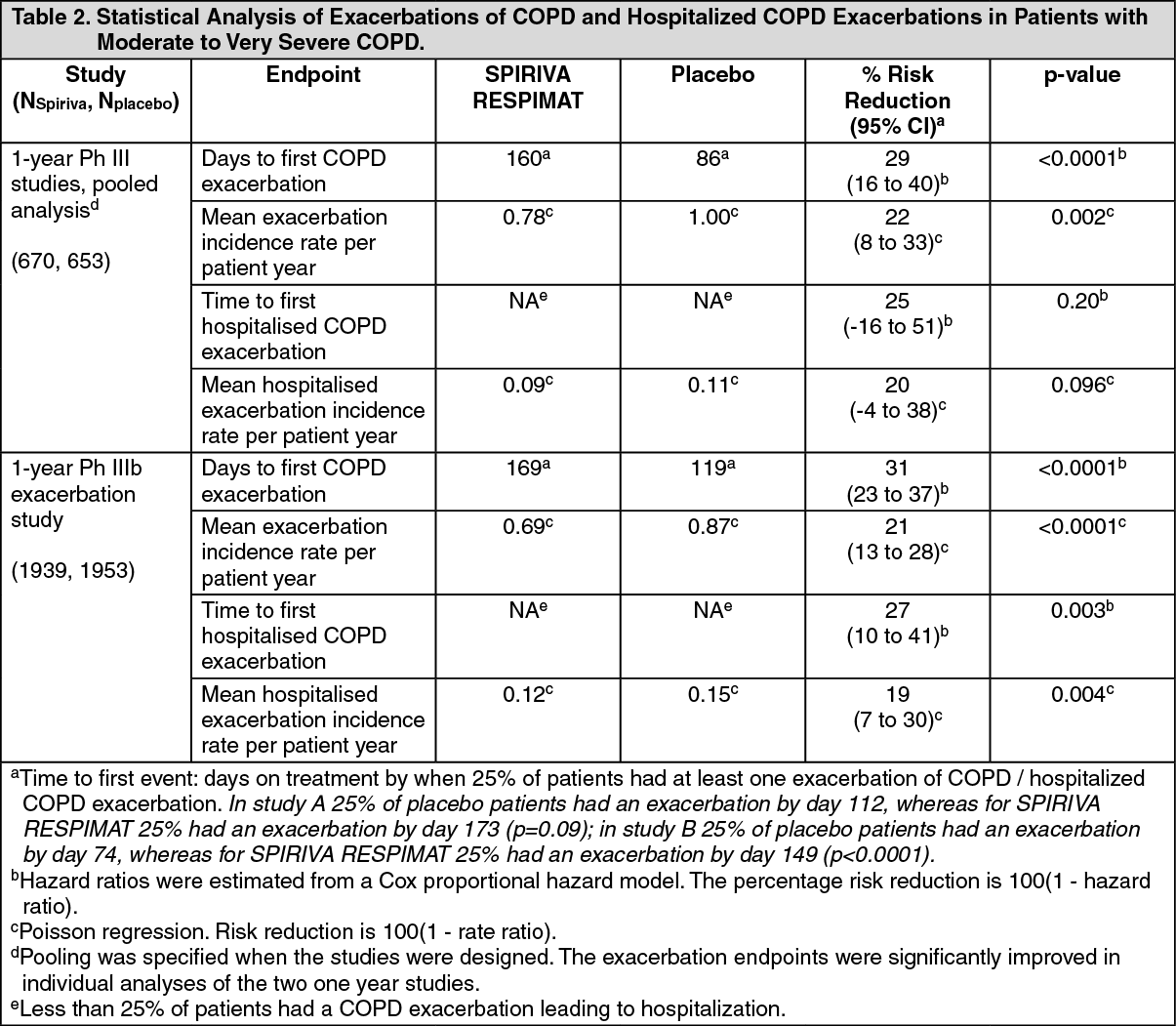
(c) COPD Exacerbations: In three one-year, randomised, double-blind, placebo-controlled clinical trials SPIRIVA RESPIMAT treatment resulted in a significantly reduced risk of a COPD exacerbation in comparison to placebo. Exacerbations of COPD were defined as "a complex of at least two respiratory events/symptoms with a duration of three days or more requiring a change in treatment (prescription of antibiotics and/or systemic corticosteroids and/or a significant change of the prescribed respiratory medication)". SPIRIVA RESPIMAT treatment resulted in a reduced risk of a hospitalisation due to a COPD exacerbation (significant in the appropriately powered large exacerbation trial).
The pooled analysis of two Phase III trials and separate analysis of an additional exacerbation trial is displayed in Table 2. All respiratory medications except anticholinergics and long-acting beta-agonists were allowed as concomitant treatment, i.e. rapidly acting beta-agonists, inhaled corticosteroids and xanthines. Long-acting beta-agonists were allowed in addition in the exacerbation trial. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageLong-term tiotropium active- controlled study: A long term, large scale, randomised, double-blind, active-controlled study with an observation period up to 3 years has been performed to compare the efficacy and safety of SPIRIVA RESPIMAT and Spiriva HandiHaler (5,711 patients receiving SPIRIVA RESPIMAT 2.5 microgram (2 puffs comprise one medicinal dose of 5 micrograms); 5,694 patients receiving Spiriva HandiHaler). The primary endpoints were time to first COPD exacerbation, time to all-cause mortality and in a sub-study (906 patients) trough FEV1 (pre-dose).
The time to first COPD exacerbation was similar during the study with SPIRIVA RESPIMAT and Spiriva HandiHaler (hazard ratio (SPIRIVA RESPIMAT / Spiriva HandiHaler) 0.98 with a 95% CI of 0.93 to 1.03).
The median number of days to the first COPD exacerbation was 756 days for SPIRIVA RESPIMAT and 719 days for Spiriva HandiHaler.
The bronchodilator effect of SPIRIVA RESPIMAT was sustained over 120 weeks, and was similar to Spiriva HandiHaler. The mean difference in trough FEV1 for SPIRIVA RESPIMAT versus Spiriva HandiHaler was -0.010 L (95% CI -0.038 to 0.018 L).
All-cause mortality was similar during the study with SPIRIVA RESPIMAT and SPIRIVA HandiHaler (hazard ratio (SPIRIVA RESPIMAT / Spiriva HandiHaler) 0.96 with a 95% CI of 0.84 to 1.09).
Asthma: Adult Patients: The clinical Phase III programme for persistent asthma included two 48 week, two 6-month and one 12-week, randomised, double-blind, placebo-controlled studies in a total of 3,476 asthma patients (1,128 receiving SPIRIVA RESPIMAT, tiotropium 5 microgram, once daily) on background treatment of at least ICS or ICS/LABA. The two 6-month studies were also active-controlled (salmeterol). All 5 studies included lung function measurements, assessments of symptoms including exacerbations, and health-related quality of life.
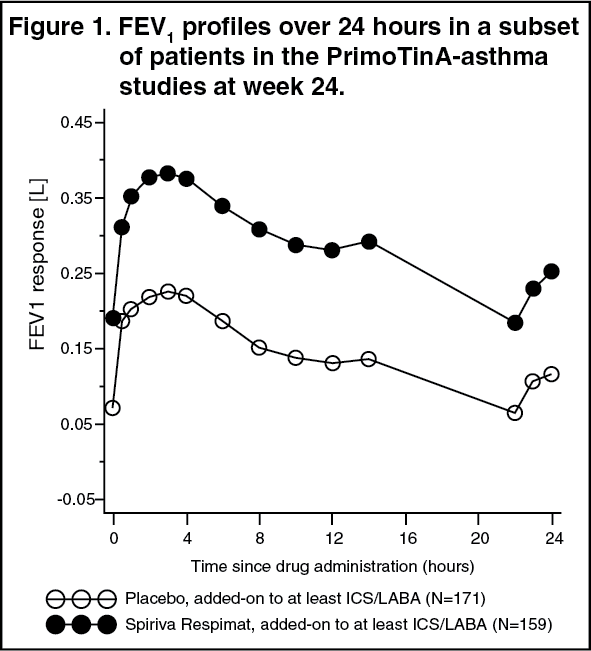
In the two 48-week PrimoTinA-asthma studies in patients who were symptomatic on maintenance treatment of at least high-dose ICS plus LABA, SPIRIVA RESPIMAT showed significant improvements in lung function over placebo when used as add-on to background treatment.
At week 24, mean improvements in peak and trough FEV1 were 0.110 litres (95% CI: 0.063 to 0.158 litres, p<0.0001) and 0.093 litres (95% CI: 0.050 to 0.137 litres, p<0.0001), respectively.
The improvement of lung function compared to placebo was maintained for 24 hours (see Figure 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAt week 24, SPIRIVA RESPIMAT significantly improved morning and evening peak expiratory flow (PEF; mean improvement in the morning 23 L/min; 95% CI: 16 to 29 L/min, p< 0.0001; evening 26 L/min; 95% CI: 20 to 33 L/min, p<0.0001).
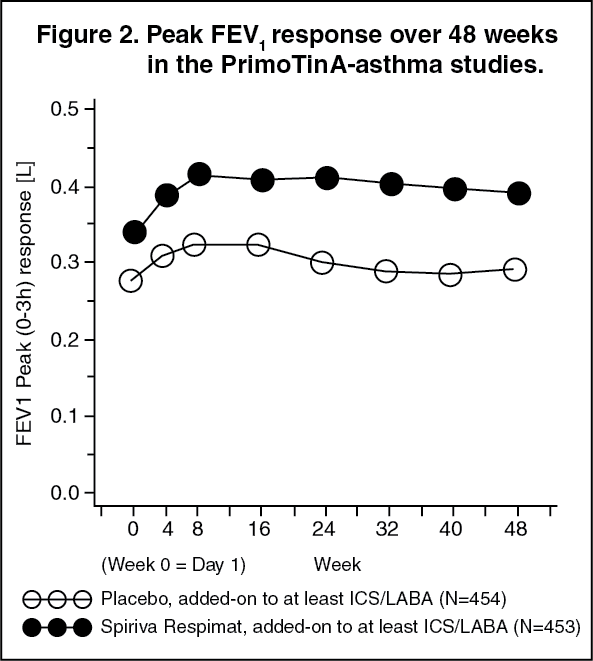
The bronchodilator effects of SPIRIVA RESPIMAT were maintained throughout the 48-week period of administration with no evidence of tachyphylaxis or tolerance (see Figure 2).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSPIRIVA RESPIMAT significantly reduced the risk of severe asthma exacerbations (see Table 3 and Figure 3).
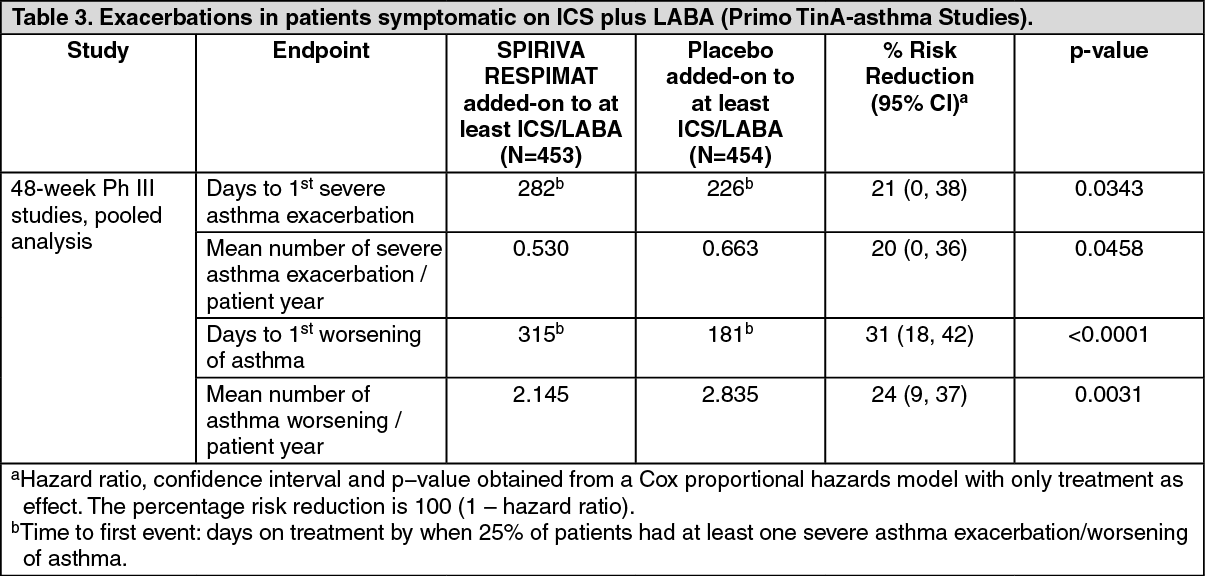 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image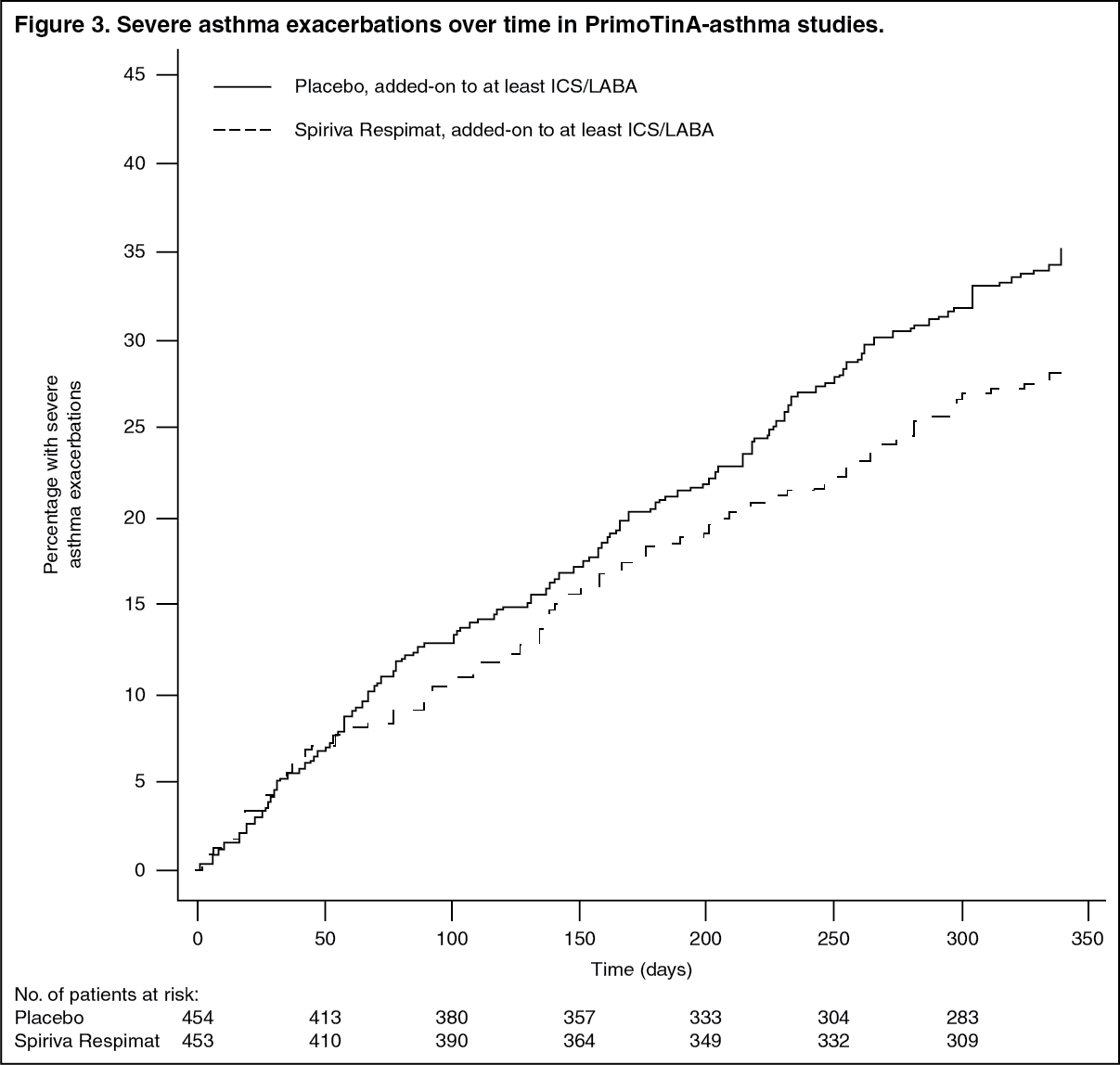 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe Asthma Control Questionnaire (ACQ) responder rates, defined as percentage of patients improving by at least 0.5 points, were significantly higher with SPIRIVA RESPIMAT (53.9% versus 46.9%; p=0.0427).
The Asthma Quality of Life Questionnaire (AQLQ(S)) mean scores for SPIRIVA RESPIMAT improved significantly over placebo at week 24 (treatment difference: 0.117, 95% CI: 0.011, 0.223, p=0.0312).
In the two 6-month MezzoTinA-asthma studies in patients who were symptomatic on maintenance treatment of medium-dose ICS, SPIRIVA RESPIMAT showed significant improvements in lung function over placebo when used as add-on to background treatment.
At week 24, mean improvements in peak and trough FEV1 were 0.185 litres (95% CI: 0.146 to 0.223 litres, p<0.0001) and 0.146 litres (0.105 to 0.188 litres, p<0.0001), respectively. The peak and trough FEV1 values for salmeterol were 0.196 litres (95% CI: 0.158 to 0.234 litres) and 0.114 litres (95% CI: 0.073 to 0.155 litres), respectively.
SPIRIVA RESPIMAT significantly improved morning and evening PEF (morning 24 L/min; 95% CI: 18 to 31 L/min, p< 0.0001; evening 23 L/min; 95% CI: 17 to 30 L/min, p<0.0001). The morning and evening PEF for salmeterol compared to placebo were 25 L/min (95% CI: 19 to 31 L/min) and 21 L/min (95% CI: 15 to 27 L/min), respectively.
Patients who took SPIRIVA RESPIMAT had a significantly higher ACQ responder rate at week 24 compared to patients taking placebo (see Table 4).
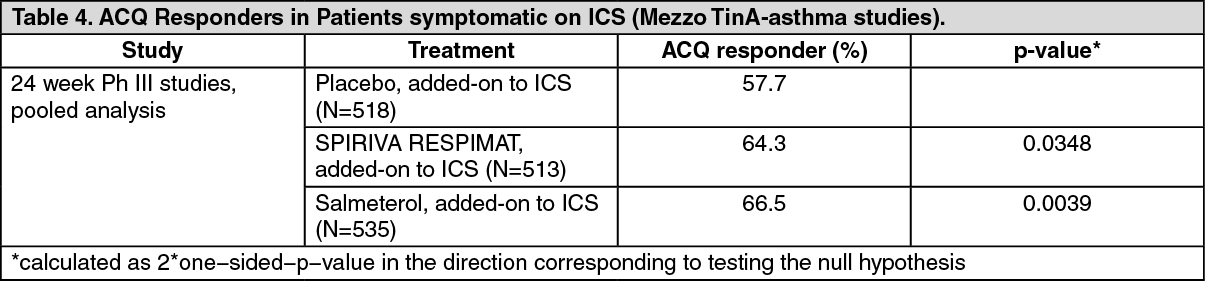 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the 12 week GraziaTinA-asthma study in patients who were symptomatic on maintenance treatment with low dose ICS, SPIRIVA RESPIMAT showed significant improvements in lung function over placebo when used as add-on to background treatment. At 12 weeks, the mean improvements in peak and trough FEV1 were 0.128 litres (95% CI: 0.057 to 0.199 litres, p<0.0005) and 0.122 litres (95% CI: 0.049 to 0.194 litres, p<0.0010), respectively.
Paediatric Patients: The clinical phase III program for persistent asthma in paediatric patients (1-17 years) was based on the following clinical trials and a partial extrapolation of data from adults: Adolescents (12-17 years): one 1-year and one 12-week randomised, double-blind, placebo-controlled studies in a total of 789 asthma patients (264 receiving SPIRIVA RESPIMAT, tiotropium 5 microgram, once daily).
Children (6-11 years): one 1-year and one 12-week randomised, double-blind, placebo-controlled studies in a total of 801 asthma patients (265 receiving SPIRIVA RESPIMAT tiotropium 5 microgram, once daily).
Children (1-5 years): one 12-week randomised, double-blind placebo-controlled study in a total of 101 asthma patients (31 receiving SPIRIVA RESPIMAT tiotropium 5 microgram, once daily).
In all these studies, patients were on background treatment of at least ICS.
Adolescents (12-17 years): In the 1-year RubaTinA-asthma study in patients with moderate asthma who were symptomatic on maintenance treatment of at least medium-dose ICS, SPIRIVA RESPIMAT showed significant improvements in lung function over placebo when used as add-on to background treatment.
At week 24, mean improvements in peak and trough FEV1 were 0.174 litres (95% CI: 0.076 to 0.272 litres, p=0.0005) and 0.117 litres (95% CI: 0.010 to 0.223 litres, p=0.0320), respectively.
At week 24, SPIRIVA RESPIMAT significantly improved morning and evening PEF (morning 15.8 L/min; 95% CI: 2.3, 29.3 L/min, p=0.0214; evening 16.7 L/min; 95% CI: 3.4, 30.0 L/min, p=0.0137).
The bronchodilator effects of SPIRIVA RESPIMAT were maintained throughout the 1 year period of administration with no evidence of tachyphylaxis (see Figure 4).
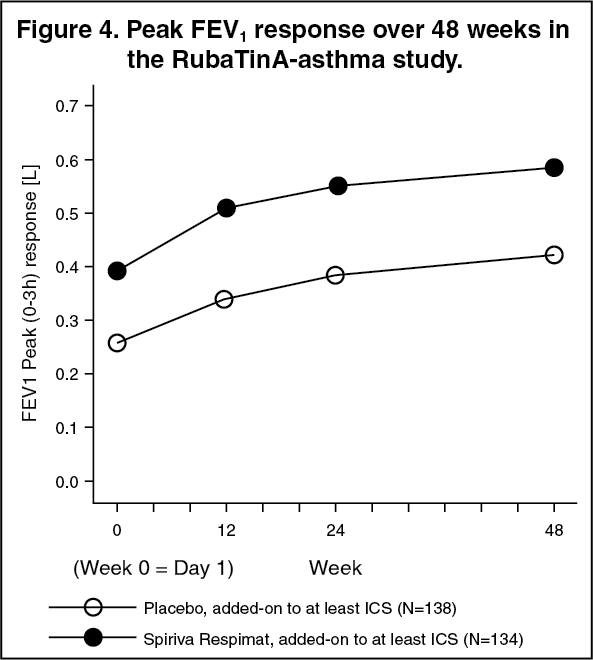 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the 12-week PensieTinA-asthma study in patients with severe asthma who were symptomatic on maintenance treatment of at least medium dose ICS in combination with 1 or more controller medication (e.g. LABA), SPIRIVA RESPIMAT showed improvements in lung function over placebo when used as add-on to background treatment, however, the differences in peak and trough FEV1 were not statistically significant.
At week 12, mean improvements in peak and trough FEV1 were 0.090 litres (95% CI: - 0.019 to 0.198 litres, p=0.1039) and 0.054 litres (95% CI: -0.061 to 0.168 litres, p=0.3605), respectively.
At week 12, SPIRIVA RESPIMAT significantly improved morning and evening PEF (morning 17.4 L/min; 95% CI: 5.1 to 29.6 L/min; evening 17.6 L/min; 95% CI: 5.9 to 29.6 L/min).
Children (6-11 years): In the 1-year CanoTinA-asthma study in patients with moderate asthma who were symptomatic on maintenance treatment of at least medium-dose ICS, SPIRIVA RESPIMAT showed significant improvements in lung function over placebo when used as add-on to background treatment.
At week 24, mean improvements in peak and trough FEV1 were 0.164 litres (95% CI: 0.103 to 0.225 litres, p<0.0001) and 0.118 litres (95% CI: 0.048 to 0.188 litres, p=0.0010), respectively.
The bronchodilator effects of SPIRIVA RESPIMAT were maintained throughout the 1 year period of administration with no evidence of tachyphylaxis (see Figure 5).
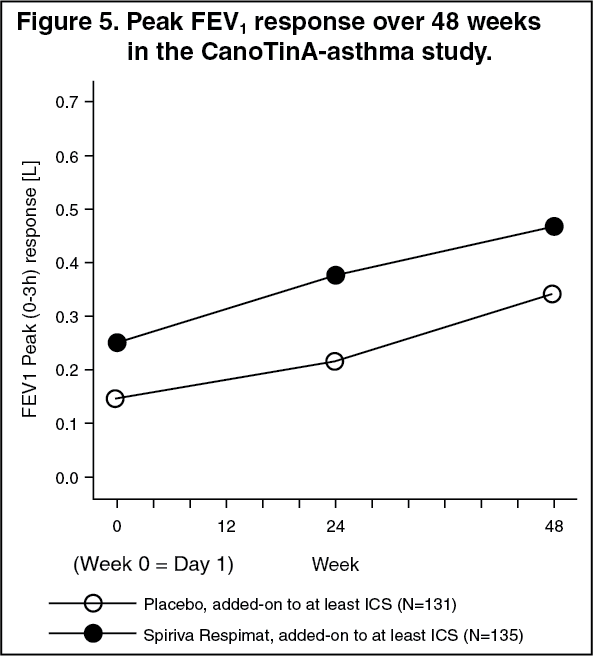 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the 12-week VivaTinA-asthma study in patients with severe asthma who were symptomatic on maintenance treatment of at least medium dose ICS in combination with 1 or more controller medication (e.g. LABA), SPIRIVA RESPIMAT showed significant improvements in lung function over placebo when used as add-on to background treatment.
At week 12, mean improvements in peak and trough FEV1 were 0.139 litres (95% CI: 0.075 to 0.203 litres, p<0.0001) and 0.087 litres (95% CI: 0.019 to 0.154 litres, p=0.0117), respectively.
Pharmacokinetics: Spiriva: General Introduction: Tiotropium bromide is a non-chiral quaternary ammonium compound and is sparingly soluble in water. Tiotropium bromide is administered by dry powder inhalation. Generally with the inhaled route of administration, the majority of the delivered dose is deposited in the gastro-intestinal tract, and to a lesser extent in the intended organ of the lung. Many of the pharmacokinetic data described as follows were obtained with higher doses than recommended for therapy.
General Characteristics of the Active Substance after Administration of the Medicinal Product: Absorption: Following dry powder inhalation by young healthy volunteers, the absolute bioavailability of 19.5% suggests that the fraction reaching the lung is highly bioavailable. Oral solutions of tiotropium have an absolute bioavailability of 2-3%.
Maximum tiotropium plasma concentrations were observed 5-7 minutes after inhalation. At steady state, peak tiotropium plasma levels in COPD patients were 12.9 pg/ml and decreased rapidly in a multi-compartmental manner. Steady state trough plasma concentrations were 1.71 pg/ml. Systemic exposure following the inhalation of tiotropium via the HandiHaler device was similar to tiotropium inhaled via the Respimat inhaler.
Distribution: Tiotropium has a plasma protein binding of 72% and shows a volume of distribution of 32 L/kg. Local concentrations in the lung are not known, but the mode of administration suggests substantially higher concentrations in the lung. Studies in rats have shown that tiotropium bromide does not penetrate the blood-brain barrier to any relevant extent.
Biotransformation: The extent of biotransformation is small. This is evident from a urinary excretion of 74% of unchanged substance after an intravenous dose to young healthy volunteers. The ester tiotropium bromide is nonenzymatically cleaved to the alcohol (N-methylscopine) and acid compound (dithienylglycolic acid) that are inactive on muscarinic receptors. In-vitro experiments with human liver microsomes and human hepatocytes suggest that some further drug (< 20% of dose after intravenous administration) is metabolised by cytochrome P450 (CYP) dependent oxidation and subsequent glutathion conjugation to a variety of Phase II-metabolites.
In vitro studies in liver microsomes reveal that the enzymatic pathway can be inhibited by the CYP 2D6 (and 3A4) inhibitors, quinidine, ketoconazole and gestodene. Thus CYP 2D6 and 3A4 are involved in metabolic pathway that is responsible for the elimination of a smaller part of the dose. Tiotropium bromide even in supra-therapeutic concentrations does not inhibit CYP 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 or 3A in human liver microsomes.
Elimination: The effective half-life of tiotropium ranges between 27-45 h in COPD patients. Total clearance was 880 ml/min after an intravenous dose in young healthy volunteers. Intravenously administered tiotropium is mainly excreted unchanged in urine (74%). After dry powder inhalation by COPD patients to steady-state, urinary excretion is 7% (1.3 μg) of the unchanged drug over 24 hours, the remainder being mainly non-absorbed drug in gut that is eliminated via the faeces. The renal clearance of tiotropium exceeds the creatinine clearance, indicating secretion into the urine. After chronic once daily inhalation by COPD patients, pharmacokinetic steady state was reached by day 7 with no accumulation thereafter.
Linearity / Nonlinearity: Tiotropium demonstrates linear pharmacokinetics in the therapeutic range independent of the formulation.
Characteristics in Patients: Geriatric Patients: As expected for all predominantly renally excreted drugs, advancing age was associated with a decrease of tiotropium renal clearance (365 mL/min in COPD patients < 65 years to 271 mL/min in COPD patients ≥ 65 years). This did not result in a corresponding increase in AUC0-6, ss or Cmax,ss values.
Renally Impaired Patients: Following once daily inhaled administrations of tiotropium to steady-state in COPD patients, mild renal impairment (CLCR 50-80 ml/min) resulted in slightly higher AUC0-6,ss (between 1.8-30% higher) and similar Cmax, ss values compared to patients with normal renal function(CLCR >80 ml/min).
In COPD patients with moderate to severe renal impairment (CLCR <50 ml/min), the intravenous administration of tiotropium resulted in doubling of the total exposure (82% higher AUC0-4h) and 52% higher Cmax) compared to COPD patients with normal renal function, which was confirmed by plasma concentrations after dry powder inhalation.
Hepatically Impaired Patients: Liver insufficiency is not expected to have any relevant influence on tiotropium pharmacokinetics. Tiotropium is predominantly cleared by renal elimination (74% in young healthy volunteers) and simple non-enzymatic ester cleavage to pharmacologically inactive products.
Japanese COPD Patients: In cross trial comparison, mean peak tiotropium plasma concentrations 10 minutes post-dosing at steady-state were 20% to 70% higher in Japanese compared to Caucasian COPD patients following inhalation of tiotropium but there was no signal for higher mortality or cardiac risk in Japanese patients compared to Caucasian patients. Insufficient pharmacokinetic data is available for other ethnicities or races.
Paediatric Patients: See Dosage & Administration.
Pharmacokinetic / Pharmacodynamic Relationship(s): There is no direct relationship between pharmacokinetics and pharmacodynamics.
Spiriva Respimat: Tiotropium bromide is a non-chiral quaternary ammonium compound and is sparingly soluble in water. Tiotropium bromide is available as solution for inhalation administered by the RESPIMAT inhaler. Generally with the inhaled route of administration, the majority of the delivered dose is swallowed and deposited in the gastrointestinal tract, and to a lesser extent is delivered to the lungs. Approximately 40% of the inhaled dose of tiotropium RESPIMAT is deposited in the lungs, the target organ, the remaining amount being deposited in the gastrointestinal tract. Some of the tiotropium RESPIMAT pharmacokinetic data described as follows were obtained with higher doses than recommended for therapy.
Bioequivalence: The primary objective of the Phase II, crossover study 205.458 involving 123 patients with COPD was to compare the pharmacokinetics of 5 μg tiotropium solution for inhalation delivered by the RESPIMAT Inhaler (Tio R 5) with tiotropium powder for inhalation 18 μg delivered by the HandiHaler (Tio HH 18). The exposure to tiotropium following the use of Tio R 5 was lower compared to Tio HH 18. Using the parameters AUC0‐6,ss and Cmax,ss, bioequivalence was not established between Tio R 5 and Tio HH 18. The ratio of AUC0‐6,ss (Tio R 5/ Tio HH 18) was 75.99% (90% confidence interval of (70.44, 81.98)). The ratio of Cmax,ss was 80.66% (90% CI: 73.49, 88.52).
Absorption: Following inhalation by young healthy volunteers, urinary excretion data suggests that approximately 33% of the inhaled dose reaches the systemic circulation. It is expected from the chemical structure of the compound that tiotropium is poorly absorbed from the gastro-intestinal tract. This was confirmed in a study in young healthy volunteers, with a low bioavailability of 2-3% for oral solutions. Food is not expected to influence the absorption of tiotropium for the same reason. Maximum tiotropium plasma concentrations were observed 5-7 minutes after inhalation. At steady state, peak tiotropium plasma concentrations of 10.5 pg/mL were achieved in patients with COPD and decreased rapidly in a multi-compartmental manner. Steady state trough plasma concentrations were 1.60 pg/mL. A steady state tiotropium peak plasma concentration of 5.15 pg/mL was attained 5 minutes after the administration of the same dose to patients with asthma.
Distribution: The drug has a plasma protein binding of 72% and shows a volume of distribution of 32 L/kg.
Local concentrations in the lung are not known, but the mode of administration suggests substantially higher concentrations in the lung. Studies in rats have shown that tiotropium does not penetrate the blood-brain barrier to any relevant extent.
Metabolism: Metabolism does not occur to any great extent in young healthy volunteers, as indicated by 74% renal excretion of unchanged drug after an intravenous dose. The major metabolic pathway is non-enzymatic ester cleavage to the alcohol N-methylscopine and dithienylglycolic acid that are inactive on muscarinic receptors.
In vitro metabolism: In studies in animals and in vitro experiments with human liver microsomes and hepatocytes, minor amounts of a variety of glutathione conjugates, after oxidation of the thiophene rings, were observed.
In vitro studies in human liver microsomes revealed that the enzymatic pathway, relevant for only a small amount of tiotropium metabolism, can be inhibited by cytochrome P450 (CYP) 2D6 inhibitor quinidine and CYP 3A4 inhibitors ketoconazole and gestodene.
Tiotropium, even in supra-therapeutic concentrations, does not inhibit CYP 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 or 3A in human liver microsomes.
Excretion: The effective half-life of tiotropium ranges between 27 to 45 h following inhalation by patients with COPD or asthma.
The effective half-life was 34 hours in patients with asthma.
Total clearance was 880 mL/min after an intravenous dose in young healthy volunteers. Urinary excretion of unchanged substance in young healthy volunteers is 74% of an intravenous dose. After inhalation of the solution for inhalation by patients with COPD, urinary excretion is 18.6% (0.93 μg) of the dose, the remainder being mainly non-absorbed drug in gut that is eliminated via the faeces.
In patients with asthma, 11.9% (0.595 μg) of the dose is excreted unchanged in the urine over 24 hours post dose at steady state.
The renal clearance of tiotropium exceeds the creatinine clearance, indicating secretion into the urine. After chronic, once daily inhalation, pharmacokinetic steady state was reached by day 7, with no accumulation thereafter.
Tiotropium demonstrates linear pharmacokinetics in the therapeutic range, independent of the formulation.
Special populations: Elderly patients: As expected for all predominantly renally excreted drugs, advancing age was associated with a decrease of tiotropium renal clearance from 347 mL/min in patients with COPD <65 years to 275 mL/min in patients with COPD ≥ 65 years. This did not result in a corresponding increase in AUC0-6,ss and Cmax,ss values.
Exposure to tiotropium was not found to differ with age in patients with asthma.
Paediatric Patients: The peak and total exposure to tiotropium was not found to differ between paediatric patients (aged 6 to 17 years) and adults with asthma. In patients 1 to 5 years old with asthma (n=3), the total exposure as measured by urinary excretion (over 3 hours) was 52 to 60% lower than that observed in patients 6 years and older with asthma; the total exposure data when adjusted for body surface area were found to be comparable in all age groups. SPIRIVA RESPIMAT was administered with a valved holding chamber with facemask in patients 1 to 5 years of age.
Renally impaired patients: Following once daily inhaled administration of tiotropium to steady-state to patients with COPD with mild renal impairment (CLCR 50-80 mL/min) resulted in slightly higher AUC0-6,ss (between 1.8 to 30% higher) and similar Cmax,ss compared to COPD patients with normal renal function (CLCR >80 mL/min. In patients with COPD with moderate to severe renal impairment (CLCR <50 mL/min), the intravenous administration of tiotropium resulted in a doubling of the total exposure (82% higher AUC0-4h and 52% higher Cmax) compared to patients with COPD with normal renal function, which was confirmed by plasma concentrations after dry powder inhalation.
In asthma patients with mild renal impairment (CLCR 50-80 mL/min) inhaled tiotropium did not result in relevant increases in exposure compared to patients with normal renal function.
Hepatically impaired patients: There are no data on the pharmacokinetics of tiotropium in hepatic impairment. Liver insufficiency is not expected to have any relevant influence on tiotropium pharmacokinetics. Tiotropium is predominantly cleared by renal elimination (74% in young healthy volunteers) and by simple non-enzymatic ester cleavage to products that do not bind to muscarinic receptors.
Toxicology: Spiriva: Preclinical safety data: Many effects observed in conventional studies of safety pharmacology, repeated dose toxicity, and reproductive toxicity could be explained by the anticholinergic properties of tiotropium bromide. Typically in animals reduced food consumption, inhibited body weight gain, dry mouth and nose, reduced lacrimation and salivation, mydriasis and increased heart rate were observed. Other relevant effects noted in repeated dose toxicity studies were: mild irritancy of the respiratory tract in rats and mice evinced by rhinitis and epithelial changes of the nasal cavity and larynx, and prostatitis along with proteinaceous deposits and lithiasis in the bladder in rats.
Harmful effects with respect to pregnancy, embryonal/foetal development, parturition or postnatal development could only be demonstrated at maternally toxic dose levels. Tiotropium bromide was not teratogenic in rats or rabbits. In a general reproduction and fertility study in rats, there was no indication of any adverse effect on fertility or mating performance of either treated parents or their offspring at any dosage.
The respiratory (irritation) and urogenital (prostatitis) changes and reproductive toxicity were observed at local or systemic exposures more than five-fold the therapeutic exposure. Studies on genotoxicity and carcinogenic potential revealed no special hazard for humans.