Pharmacotherapeutic group: Antivirals for systemic use, antivirals for treatment of HIV infection, combinations.
ATC code: J05AR22.
Pharmacology: Pharmacodynamics: Mechanism of action: Darunavir is an inhibitor of the dimerisation and of the catalytic activity of the HIV-1 protease (K
D of 4.5 x 10
-12M). It selectively inhibits the cleavage of HIV encoded Gag-Pol polyproteins in virus infected cells, thereby preventing the formation of mature infectious virus particles.
Cobicistat is a mechanism-based inhibitor of cytochrome P450 of the CYP3A subfamily. Inhibition of CYP3A-mediated metabolism by cobicistat enhances the systemic exposure of CYP3A substrates, such as darunavir, where bioavailability is limited and half-life is shortened due to CYP3A-dependent metabolism.
Emtricitabine is a nucleoside reverse transcriptase inhibitor (NRTI) and nucleoside analogue of 2'-deoxycytidine. Emtricitabine is phosphorylated by cellular enzymes to form emtricitabine triphosphate. Emtricitabine triphosphate inhibits HIV replication through incorporation into viral DNA by the HIV reverse transcriptase (RT), which results in DNA chain-termination.
Tenofovir alafenamide is a nucleotide reverse transcriptase inhibitor (NtRTI) and phosphonoamidate prodrug of tenofovir (2'-deoxyadenosine monophosphate analogue). Tenofovir alafenamide is permeable into cells and due to increased plasma stability and intracellular activation through hydrolysis by cathepsin A, tenofovir alafenamide is more efficient than tenofovir disoproxil in concentrating tenofovir in peripheral blood mononuclear cells (PBMC) (including lymphocytes and other HIV target cells) and macrophages. Intracellular tenofovir is subsequently phosphorylated to the pharmacologically active metabolite tenofovir diphosphate. Tenofovir diphosphate inhibits HIV replication through incorporation into viral DNA by the HIV RT, which results in DNA chain-termination.
Antiviral activity in vitro: Darunavir, emtricitabine and tenofovir alafenamide demonstrated additive to synergistic antiviral effects in two-drug combination studies in cell culture.
Darunavir exhibits activity against laboratory strains and clinical isolates of HIV-1 and laboratory strains of HIV-2 in acutely infected T-cell lines, human PBMCs and human monocytes/macrophages with median EC
50 values ranging from 1.2 to 8.5 nM (0.7 to 5.0 ng/mL). Darunavir demonstrates antiviral activity
in vitro against a broad panel of HIV-1 group M (A, B, C, D, E, F, G) and group O primary isolates with EC
50 values ranging from < 0.1 to 4.3 nM. These EC
50 values are well below the 50% cellular toxicity concentration range of 87 µM to > 100 µM.
Cobicistat has no detectable antiviral activity against HIV-1 and does not antagonise the antiviral effect of darunavir, emtricitabine, or tenofovir.
The antiviral activity of emtricitabine against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, the MAGI CCR5 cell line, and PBMCs. The EC
50 values for emtricitabine were in the range of 0.0013 to 0.64 µM. Emtricitabine displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, and G (EC
50 values ranged from 0.007 to 0.075 µM) and showed strain specific activity against HIV-2 (EC
50 values ranged from 0.007 to 1.5 µM).
The antiviral activity of tenofovir alafenamide against laboratory and clinical isolates of HIV-1 subtype B was assessed in lymphoblastoid cell lines, PBMCs, primary monocyte/macrophage cells and CD4+-T lymphocytes. The EC
50 values for tenofovir alafenamide were in the range of 2.0 to 14.7 nM. Tenofovir alafenamide displayed antiviral activity in cell culture against all HIV-1 groups (M, N, and O), including subtypes A, B, C, D, E, F, and G (EC
50 values ranged from 0.10 to 12.0 nM) and showed strain specific activity against HIV-2 (EC
50 values ranged from 0.91 to 2.63 nM).
Resistance: In vitro selection of darunavir-resistant virus from wild type HIV-1 was lengthy (> 3 years). The selected viruses were unable to grow in the presence of darunavir concentrations above 400 nM. Viruses selected in these conditions and showing decreased susceptibility to darunavir (range: 23-50-fold) harboured 2 to 4 amino acid substitutions in the protease gene. The decreased susceptibility to darunavir of the emerging viruses in the selection experiment could not be explained by the emergence of these protease mutations.
In vivo, darunavir resistance-associated mutations (V11I, V32I, L33F, I47V, I50V, I54L or M, T74P, L76V, I84V and L89V) in HIV-1 protease were derived from clinical trial data of ART-experienced patients, all of whom were protease inhibitor experienced.
Reduced susceptibility to emtricitabine is associated with M184V/I mutations in HIV-1 RT.
HIV-1 isolates with reduced susceptibility to tenofovir alafenamide express a K65R mutation in HIV-1 RT; in addition, a K70E mutation in HIV-1 RT has been transiently observed. HIV-1 isolates with the K65R mutation have low-level reduced susceptibility to abacavir, emtricitabine, tenofovir, and lamivudine.
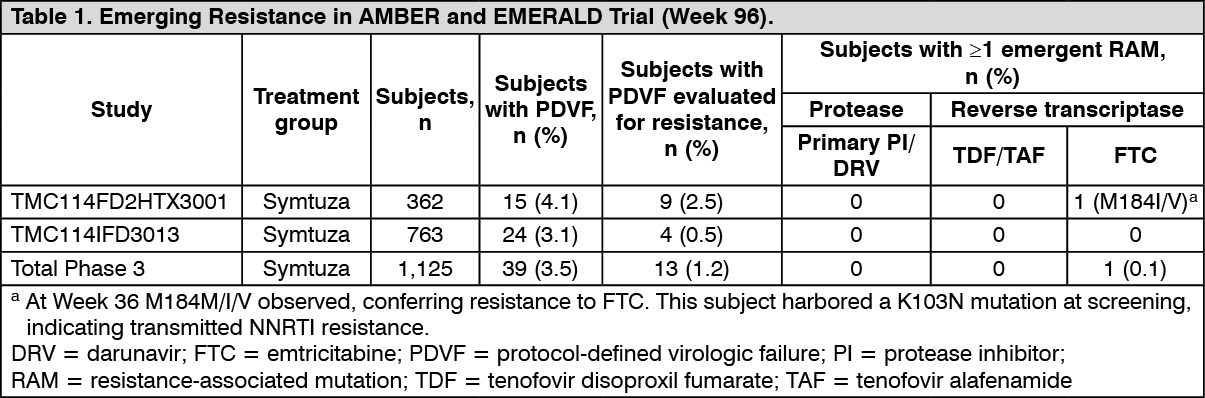
Emerging resistance in HIV-1 infected, treatment-naïve and virologically suppressed patients: Over 96 weeks of treatment in the Phase 3 studies TMC114FD2HTX3001 (AMBER) in treatment-naïve patients and TMC114IFD3013 (EMERALD) in virologically suppressed treatment-experienced patients, resistance testing was performed on samples from patients experiencing protocol-defined virologic failure (PDVF) and who had HIV-1 RNA ≥ 400 copies/mL at failure or at later time points. Emerging resistance in the Symtuza groups is shown in Table 1. No DRV, primary PI, or TDF/TAF resistance-associated mutations were observed. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Cross-resistance in HIV-1 infected, treatment-naïve
and virologically suppressed patients: The emtricitabine-resistant virus with the M184M/I/V mutation was cross-resistant to lamivudine, but retained sensitivity to abacavir, stavudine, tenofovir, and zidovudine.
Clinical data: HIV-1 Treatment-naïve Patients: In double-blind Phase 3 Trial TMC114FD2HTX3001 (AMBER), treatment-naïve patients were randomised to receive either Symtuza (N = 362) or a combination of fixed-dose combination of darunavir and cobicistat and fixed-dose combination of emtricitabine and tenofovir disoproxil fumarate (F/TDF) (N = 363) once daily. Virologic response was defined as < 50 copies/mL using the snapshot approach (see Table 2).
The 725 patients in total had a median age of 34 years (range 18-71), 88.3% were male, 83.2% White, 11.1% Black, 1.5% Asian. The mean baseline plasma HIV-1 RNA and the median baseline CD4+ cell count were 4.48 log
10 copies/mL (SD = 0.61) and 453 x 10
6 cells/L (range 38 - 1,456 x 10
6 cells/L), respectively.
(See Table 2.)
Click on icon to see table/diagram/image
Changes in measures of bone mineral density: In
the Phase 3 study TMC114FD2HTX3001 in treatment-naïve patients, Symtuza was associated with no or smaller reductions in bone mineral density (BMD) compared DRV/COBI+F/TDF as measured by DXA analysis of hip (LS means percent change: 0.17% vs -2.69%, p < 0.001) and lumbar spine (LS means percent change: -0.68% vs -2.38%, p = 0.004) after 48 weeks of treatment.
After 96 weeks of treatment with Symtuza, the (95% CI) percent changes from baseline in BMD at the hip and spine region were respectively: -0.26 (-0.96; 0.45) % and -0.93 (-1.82; -0.05) %.
Changes in measures of renal function: In studies in treatment-naïve patients, Symtuza was associated with a lower impact on the estimated glomerular filtration rate by Cockcroft-Gault method compared to control group (DRV/COBI+F/TDF).
HIV-1 Treatment-experienced Patients: Phase 3 trial TMC114IFD3013 (EMERALD) evaluated the efficacy of Symtuza in virologically-suppressed (HIV-1 RNA less than 50 copies/mL) HIV-1 infected patients. Patients were virologically suppressed for at least 2 months and no more than once had a viral load elevation above 50 HIV-1 RNA copies/mL during the year prior to enrollment. Patients were allowed in the study if they had previous failure on any non-darunavir ARV regimen. Patients had no history of virologic failure on darunavir-based regimens, and if historical genotypes were available, absence of darunavir RAMs. Patients were on a stable ARV regimen (for at least 6 months), consisting of a boosted protease inhibitor [either darunavir once daily or atazanavir (both boosted with ritonavir or cobicistat), or lopinavir with ritonavir] combined with emtricitabine and TDF. They either switched to Symtuza (N = 763) or continued their treatment regimen (N = 378) (randomised 2:1).
Patients had a median age of 46 years (range 19-78), 82% were male, 75.5% White, 20.9% Black, and 2.3% Asian. The median baseline CD4+ cell count was 628 x 10
6 cells/mm
3 (range 111-1921 x 10
6 cells/mm
3).
Week 48 and 96 virologic outcomes in the EMERALD trial are provided in Table 3. (See Table 3.)
Click on icon to see table/diagram/image
Paediatric population: The use of Symtuza in ART-naïve adolescent patients from the age of 12 years to < 18 years, and weighing at least 40 kg is supported by two trials in HIV-1 infected paediatric patients (TMC114-C230 and GS-US-292-0106). For more details, refer to the prescribing information of darunavir and emtricitabine/tenofovir alafenamide.
An open-label, Phase 2 trial (TMC114-C230) was conducted for evaluating the pharmacokinetics, safety, tolerability, and efficacy of darunavir with low dose ritonavir in 12 ART-naïve HIV-1 infected paediatric patients aged 12 to less than 18 years and weighing at least 40 kg. These patients received darunavir/ritonavir 800/100 mg once daily in combination with other antiretroviral agents. Virologic response was defined as a decrease in plasma HIV-1 RNA viral load of at least 1.0 log10 versus baseline (see Table 4).
Click on icon to see table/diagram/image
In the study GS-US-292-0106, the efficacy, safety, and pharmacokinetics of emtricitabine and tenofovir alafenamide were evaluated in an open-label study in which 50 HIV-1 infected, treatment-naïve adolescents received emtricitabine and tenofovir alafenamide (10 mg) given with elvitegravir and cobicistat as a fixed-dose combination tablet. Patients had a median age of 15 years (range: 12-17), and 56% were female, 12% were Asian, and 88% were Black. At baseline, median plasma HIV-1 RNA was 4.7 log10 copies/mL, median CD4+ cell count was 456 cells/mm3 (range: 95-1,110), and median CD4+ % was 23% (range: 7-45%). Overall, 22% had baseline plasma HIV-1 RNA > 100,000 copies/mL. At 48 weeks, 92% (46/50) achieved HIV-1 RNA < 50 copies/mL, similar to response rates in studies of treatment-naïve HIV-1 infected adults. The mean increase from baseline in CD4+ cell count at Week 48 was 224 cells/mm3. No emergent resistance to E/C/F/TAF (elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide) was detected through Week 48.
The European Medicines Agency has deferred the obligation to submit the results of studies with Symtuza in one or more subsets of the paediatric population in the treatment of HIV-1 infection (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: The bioavailability of all components of Symtuza was comparable to that when darunavir 800 mg, cobicistat 150 mg, and emtricitabine/tenofovir alafenamide 200/10 mg were co-administered as separate formulations; bioequivalence was established following single-dose administration under fed conditions in healthy subjects (N = 96).
Absorption: The absolute bioavailability of a single 600 mg dose of darunavir alone was approximately 37% and increased to approximately 82% in the presence of 100 mg twice daily ritonavir. The absolute bioavailability of the emtricitabine 200 mg capsule was 93%.
All components were rapidly absorbed following oral administration of Symtuza in healthy subjects. Maximum plasma concentrations of darunavir, cobicistat, emtricitabine and tenofovir alafenamide were achieved at 4.00, 4.00, 2.00, and 1.50 hours after dosing, respectively. The bioavailability of the components of Symtuza was not affected when administered orally as a split tablet compared to administration as a tablet swallowed whole.
The exposure to darunavir and cobicistat administered as the Symtuza was 30-45% lower and 16-29% lower, respectively, in fasted compared to fed condition. For emtricitabine, the C
max was 1.26-fold higher in a fasted condition, while the AUC was comparable in fed and fasted condition. For tenofovir alafenamide, the C
max was 1.82-fold higher in fasted condition, while the AUC was 20% lower to comparable in a fasted compared to fed condition. Symtuza tablets should be taken with food. The type of food does not affect exposure to Symtuza.
Distribution: Darunavir: Darunavir is approximately 95% bound to plasma protein. Darunavir binds primarily to plasma α
1-acid glycoprotein.
Following intravenous administration, the volume of distribution of darunavir alone was 88.1 ± 59.0 L (mean ± SD) and increased to 131 ± 49.9 L (mean ± SD) in the presence of 100 mg twice-daily ritonavir.
Cobicistat: Cobicistat is 97% to 98% bound to human plasma proteins and the mean plasma to blood-drug concentration ratio was approximately 2.
Emtricitabine:
In vitro binding of emtricitabine to human plasma proteins was < 4% and independent of concentration over the range of 0.02-200 mcg/mL. At peak plasma concentration, the mean plasma to blood drug concentration ratio was approximately 1.0 and the mean semen to plasma drug concentration ratio was approximately 4.0.
Tenofovir alafenamide:
In vitro binding of tenofovir to human plasma proteins is < 0.7% and is independent of concentration over the range of 0.01-25 mcg/mL.
Ex vivo binding of tenofovir alafenamide to human plasma proteins in samples collected during clinical studies was approximately 80%.
Biotransformation: Darunavir:
In vitro experiments with human liver microsomes (HLMs) indicate that darunavir primarily undergoes oxidative metabolism. Darunavir is extensively metabolised by the hepatic CYP system and almost exclusively by isozyme CYP3A4. A [
14C]-darunavir trial in healthy volunteers showed that a majority of the radioactivity in plasma after a single 400/100 mg darunavir with ritonavir dose was due to the parent active substance. At least 3 oxidative metabolites of darunavir have been identified in humans; all showed activity that was at least 10-fold less than the activity of darunavir against wild type HIV.
Cobicistat: Cobicistat is metabolised via CYP3A (major)- and CYP2D6 (minor)-mediated oxidation and does not undergo glucuronidation. Following oral administration of [
14C]-cobicistat, 99% of circulating radioactivity in plasma was unchanged cobicistat. Low levels of metabolites are observed in urine and faeces and do not contribute to the CYP3A inhibitory activity of cobicistat.
Emtricitabine:
In vitro studies indicate that emtricitabine is not an inhibitor of human CYP enzymes. Following administration of [
14C]-emtricitabine, complete recovery of the emtricitabine dose was achieved in urine (approximately 86%) and faeces (approximately 14%). Thirteen percent of the dose was recovered in the urine as three putative metabolites. The biotransformation of emtricitabine includes oxidation of the thiol moiety to form the 3'-sulfoxide diastereomers (approximately 9% of dose) and conjugation with glucuronic acid to form 2'-O-glucuronide (approximately 4% of dose). No other metabolites were identifiable.
Tenofovir alafenamide: Metabolism is a major elimination pathway for tenofovir alafenamide in humans, accounting for > 80% of an oral dose.
In vitro studies have shown that tenofovir alafenamide is metabolised to tenofovir (major metabolite) by cathepsin A in PBMCs (including lymphocytes and other HIV target cells) and macrophages; and by carboxylesterase-1 in hepatocytes.
In vivo, tenofovir alafenamide is hydrolysed within cells to form tenofovir (major metabolite), which is phosphorylated to the active metabolite tenofovir diphosphate.
In vitro, tenofovir alafenamide is not metabolised by CYP1A2, CYP2C8, CYP2C9, CYP2C19, or CYP2D6. Tenofovir alafenamide is minimally metabolised by CYP3A4. Upon co-administration with the moderate CYP3A inducer probe efavirenz, tenofovir alafenamide exposure was not significantly affected. Following administration of tenofovir alafenamide, plasma [
14C]-radioactivity showed a time-dependent profile with tenofovir alafenamide as the most abundant species in the initial few hours and uric acid in the remaining period.
Elimination: Darunavir: After a 400/100 mg [
14C]-darunavir with ritonavir dose, approximately 79.5% and 13.9% of the administered dose of [
14C]-darunavir could be retrieved in faeces and urine, respectively. Unchanged darunavir accounted for approximately 41.2% and 7.7% of the administered dose in faeces and urine, respectively.
The intravenous clearance of darunavir alone (150 mg) and in the presence of low dose (100 mg) ritonavir was 32.8 l/h and 5.9 l/h, respectively. The median terminal plasma half-life of darunavir following administration of Symtuza is 5.5 hours.
Cobicistat: Following oral administration of [
14C]-cobicistat, 86% and 8.2% of the dose were recovered in faeces and urine, respectively. The median terminal plasma half-life of cobicistat following administration of Symtuza is 3.6 hours.
Emtricitabine: Emtricitabine is primarily excreted by the kidneys with complete recovery of the dose achieved in urine (approximately 86%) and faeces (approximately 14%). Thirteen percent of the emtricitabine dose was recovered in urine as three metabolites. The systemic clearance of emtricitabine averaged 307 mL/min. Following oral administration of Symtuza, the median terminal elimination half-life of emtricitabine is 17.2 hours.
Tenofovir alafenamide: Tenofovir alafenamide is mainly eliminated following metabolism to tenofovir. The median terminal elimination half-life of tenofovir alafenamide was 0.3 hours when administered as Symtuza. Tenofovir is eliminated from the body by the kidneys by both glomerular filtration and active tubular secretion. Tenofovir has a median plasma half-life of approximately 32 hours. Renal excretion of intact tenofovir alafenamide is a minor pathway with less than 1% of the dose eliminated in urine. The pharmacologically active metabolite, tenofovir diphosphate, has a half-life of 150-180 hours within PBMCs.
Special populations: Paediatric population: The pharmacokinetics of Symtuza have not been investigated in paediatric patients.
However, there are pharmacokinetic data for the different components of Symtuza, indicating that doses of 800 mg darunavir, 150 mg cobicistat, 200 mg emtricitabine and 10 mg tenofovir alafenamide result in similar exposures in adults and adolescents aged 12 years and older, weighing at least 40 kg.
Elderly: Limited PK information is available in the elderly (age ≥ 65 years of age) for Symtuza as well as its individual components.
Population pharmacokinetic analysis in HIV infected patients showed that darunavir pharmacokinetics are not considerably different in the age range (18 to 75 years) evaluated in HIV infected patients (N = 12, age ≥ 65 years) (see Precautions).
No clinically relevant pharmacokinetic differences due to age have been identified for cobicistat, emtricitabine or tenofovir alafenamide in the age range ≤ 65 years.
Gender: Population pharmacokinetic analysis showed a slightly higher darunavir exposure (16.8%) in HIV-1 infected females compared to males. This difference is not clinically relevant.
No clinically relevant pharmacokinetic differences due to gender have been identified for cobicistat, emtricitabine or tenofovir alafenamide.
Renal impairment: Symtuza has not been investigated in patients with renal impairment. There are pharmacokinetic data for the (individual) components of Symtuza.
Darunavir: Results from a mass balance study with [
14C]-darunavir with ritonavir showed that approximately 7.7% of the administered dose of darunavir is excreted in the urine unchanged.
Although darunavir has not been studied in patients with renal impairment, population pharmacokinetic analysis showed that the pharmacokinetics of darunavir were not significantly affected in HIV infected patients with moderate renal impairment (eGFR
CG between 30-60 mL/min, N = 20) (see Dosage & Administration and Precautions).
Cobicistat: A trial of the pharmacokinetics of cobicistat was performed in non-HIV-1 infected subjects with severe renal impairment (eGFR
CG below 30 mL/min). No meaningful differences in cobicistat pharmacokinetics were observed between subjects with severe renal impairment and healthy subjects, consistent with low renal clearance of cobicistat.
Emtricitabine: Mean systemic emtricitabine exposure was higher in patients with severe renal impairment (eGFR
CG < 30 mL/min) (33.7 mcg·h/mL) than in subjects with normal renal function (11.8 mcg·h/mL).
Tenofovir alafenamide: No clinically relevant differences in tenofovir alafenamide, or tenofovir pharmacokinetics were observed between healthy subjects and patients with severe renal impairment (eGFR
CG > 15 but < 30 mL/min) in studies of tenofovir alafenamide. There are no pharmacokinetic data on tenofovir alafenamide in patients with eGFR
CG < 15 mL/min.
Hepatic impairment: Symtuza has not been investigated in patients with hepatic impairment. There are pharmacokinetic data for the (individual) components of Symtuza.
Darunavir: Darunavir is primarily metabolised and eliminated by the liver. In a multiple dose trial with darunavir/ritonavir (600/100 mg) twice daily, it was demonstrated that the total plasma concentrations of darunavir in subjects with mild (Child-Pugh Class A, N = 8) and moderate (Child-Pugh Class B, N = 8) hepatic impairment were comparable with those in healthy subjects. However, unbound darunavir concentrations were approximately 55% (Child-Pugh Class A) and 100% (Child-Pugh Class B) higher, respectively. The clinical relevance of this increase is unknown. The effect of severe hepatic impairment on the pharmacokinetics of darunavir has not been studied (see Dosage & Administration, Contraindications, and Precautions).
Cobicistat: Cobicistat is primarily metabolised and eliminated by the liver. A trial of the pharmacokinetics of cobicistat was performed in non-HIV-1 infected subjects with moderate hepatic impairment (Child-Pugh Class B). No clinically relevant differences in cobicistat pharmacokinetics were observed between subjects with moderate impairment and healthy subjects. The effect of severe hepatic impairment (Child-Pugh Class C) on the pharmacokinetics of cobicistat has not been studied.
Emtricitabine: The pharmacokinetics of emtricitabine have not been studied in patients with hepatic impairment; however, emtricitabine is not significantly metabolised by liver enzymes, so the impact of liver impairment should be limited.
Tenofovir alafenamide: Clinically relevant changes in tenofovir pharmacokinetics in patients with hepatic impairment were not observed in patients with mild to moderate hepatic impairment. The effect of severe hepatic impairment (Child-Pugh Class C) on the pharmacokinetics of tenofovir alafenamide has not been studied.
Hepatitis B and/or hepatitis C virus co-infection: There were insufficient pharmacokinetic data in the clinical trials to determine the effect of hepatitis B and/or C virus infection on the pharmacokinetics of darunavir, cobicistat, emtricitabine, or tenofovir alafenamide (refer to Precautions and Adverse Reactions).
Pregnancy and postpartum: Treatment with darunavir/cobicistat 800/150 mg once daily during pregnancy results in low darunavir exposure (see Table 5). In women receiving darunavir/cobicistat during the second trimester of pregnancy, mean intra-individual values for total darunavir C
max, AUC
24h and C
min were 49%, 56% and 92% lower, respectively, as compared with postpartum; during the third trimester of pregnancy, total darunavir C
max, AUC
24h and C
min values were 37%, 50% and 89% lower, respectively, as compared with postpartum. The unbound fraction was also substantially reduced, including around 90% reductions of C
min levels. The main cause of these low exposures is a marked reduction in cobicistat exposure as a consequence of pregnancy-associated enzyme induction (see Table 5).
Click on icon to see table/diagram/image
The exposure to cobicistat was lower during pregnancy, potentially leading to suboptimal boosting of darunavir. During the second trimester of pregnancy, cobicistat C
max, AUC
24h, and C
min were 50%, 63%, and 83% lower, respectively, as compared with postpartum. During the third trimester of pregnancy, cobicistat C
max, AUC
24h, and C
min were 27%, 49%, and 83% lower, respectively, as compared with postpartum.
No pharmacokinetic data are available for emtricitabine and tenofovir alafenamide during pregnancy.
Toxicology: Preclinical safety data: Darunavir: Non-clinical data on darunavir reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential. Darunavir has no effect on fertility or early embryonic development and DRV shows no teratogenic potential, at exposure levels below those at the recommended clinical dose in humans.
In juvenile rats receiving darunavir up to days 23-26 (equivalent to less than 2 years of age in humans), increased mortality was observed with convulsions in some animals. These findings were attributed to the immaturity of the liver enzymes and of the blood brain barrier. Due to uncertainties regarding the rate of development of the human blood brain barrier and liver enzymes Symtuza should not be used in paediatric patients below 3 years of age.
Cobicistat: Non-clinical data reveal no special hazard for humans based on conventional studies of repeated dose toxicity, genotoxicity, and toxicity to reproduction and development. No teratogenic effects were observed in rats and rabbit developmental toxicity studies. In rats, ossification changes in the spinal column and sternebrae of foetuses occurred at a dose that produced significant maternal toxicity.
Ex vivo rabbit studies and
in vivo dog studies suggest that cobicistat has a low potential for QT prolongation, and may slightly prolong the PR interval and decrease left ventricular function at mean concentrations at least 10-fold higher than the human exposure at the recommended 150 mg daily dose.
A long-term carcinogenicity study of cobicistat in rats revealed tumourigenic potential specific for this species, that is regarded as of no relevance for humans. A long-term carcinogenicity study in mice did not show any carcinogenic potential.
Emtricitabine: Non-clinical data on emtricitabine reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction and development.
Emtricitabine had demonstrated low carcinogenic potential in mice and rats.
Tenofovir alafenamide: Non-clinical studies of tenofovir alafenamide in rats and dogs revealed bone and kidney as the primary target organs of toxicity. Bone toxicity was observed as reduced bone mineral density in rats and dogs at tenofovir exposures at least four times greater than those expected after administration of Symtuza. A minimal infiltration of histiocytes was present in the eye in dogs at tenofovir alafenamide and tenofovir exposures of approximately 15 and 40 times greater, respectively, than those expected after administration of Symtuza.
Tenofovir alafenamide was not mutagenic or clastogenic in conventional genotoxicity assays.
Because there is a lower tenofovir exposure in rats and mice after the administration of tenofovir alafenamide compared to tenofovir disoproxil, carcinogenicity studies and a rat peri-postnatal study were conducted only with tenofovir disoproxil. No special hazard for humans was revealed in conventional studies of carcinogenic potential and toxicity to reproduction and development. Reproductive toxicity studies in rats and rabbits showed no effects on mating, fertility, pregnancy or foetal parameters. However, tenofovir disoproxil reduced the viability index and weight of pups in a peri-postnatal toxicity study at maternally toxic doses.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image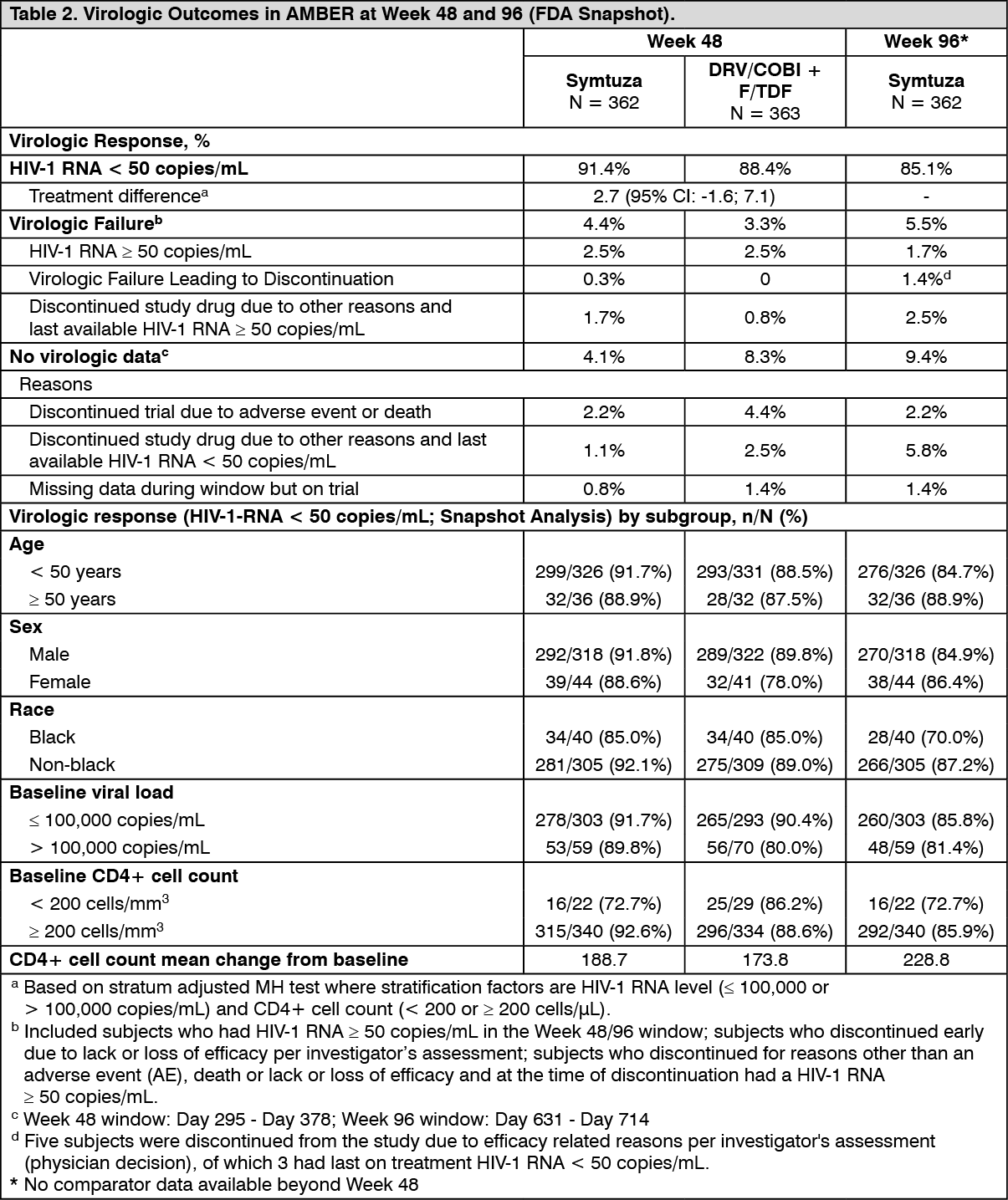 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image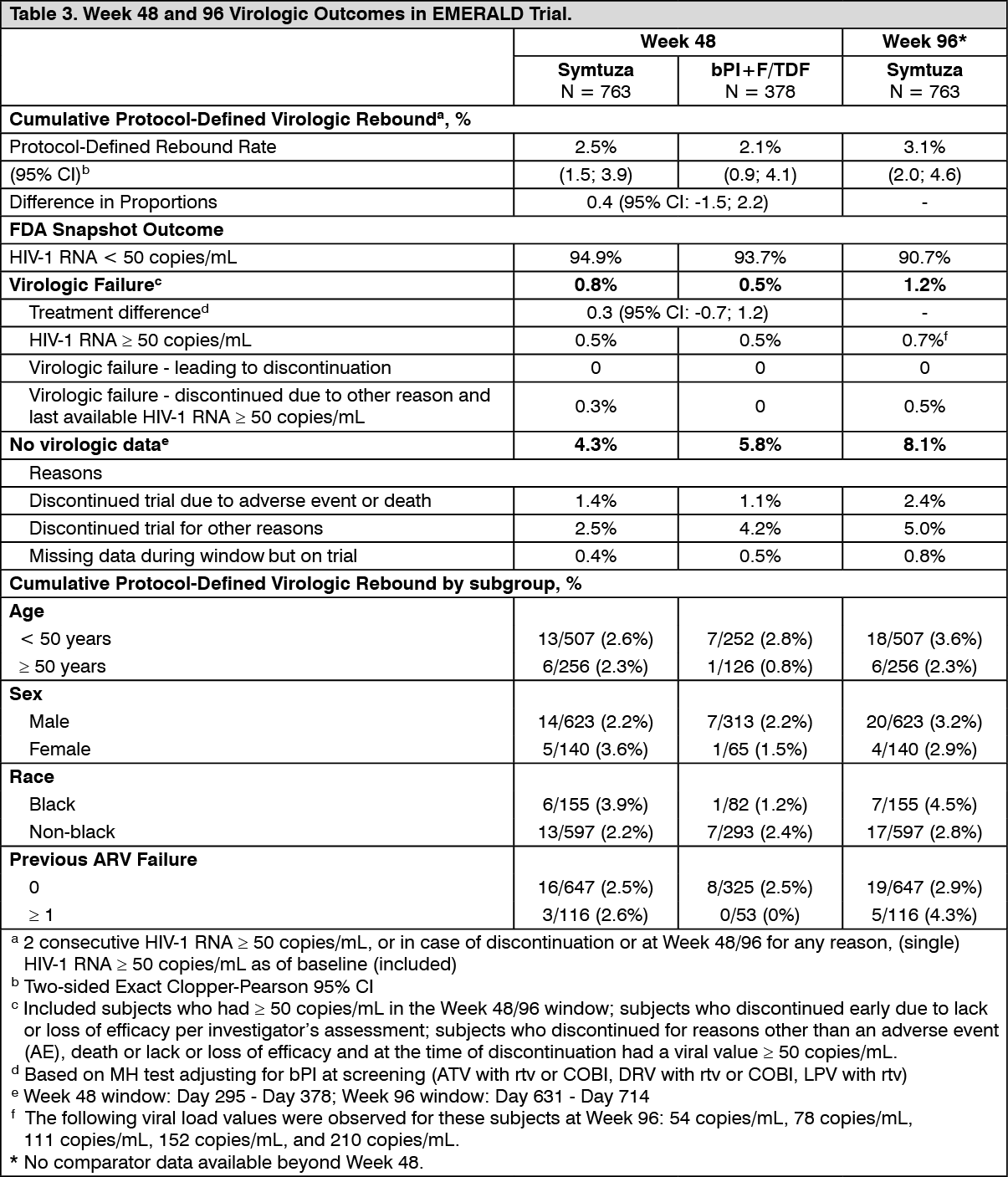 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image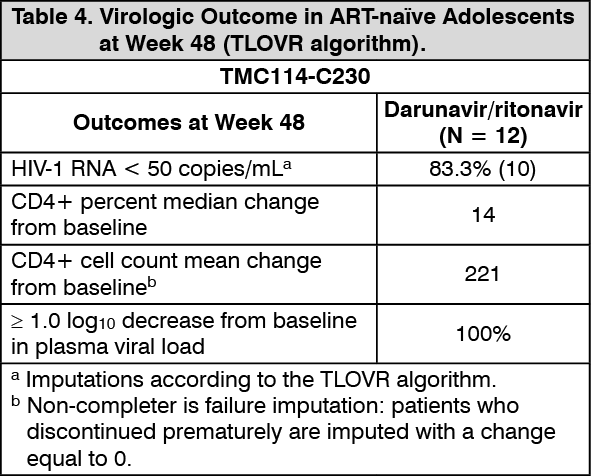 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image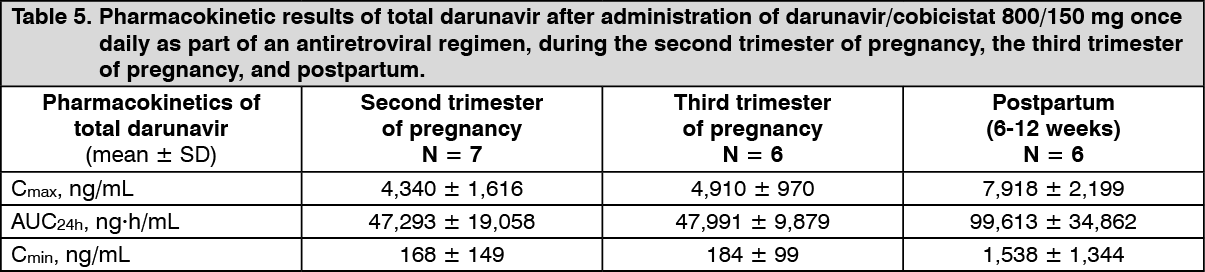 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image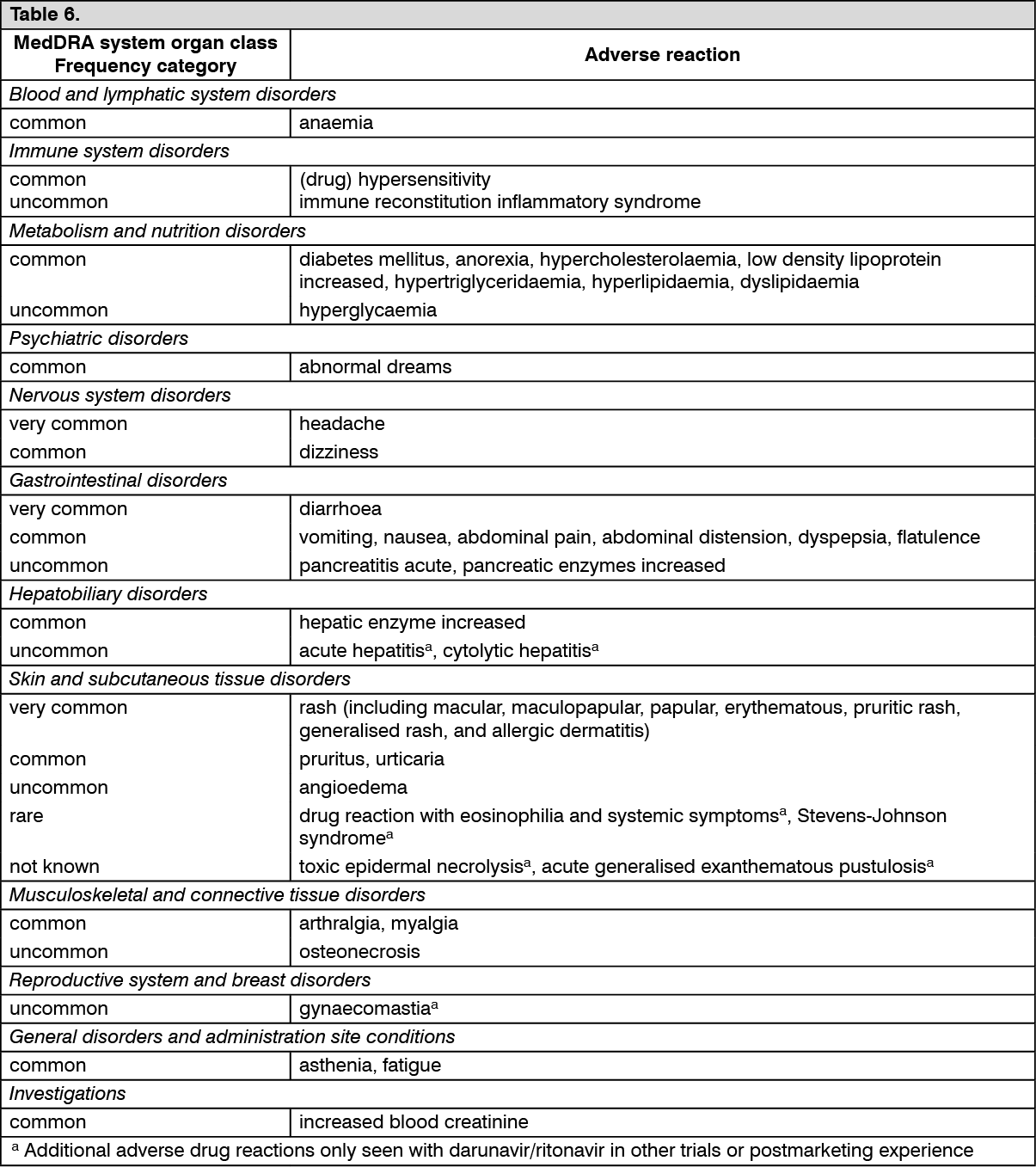 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image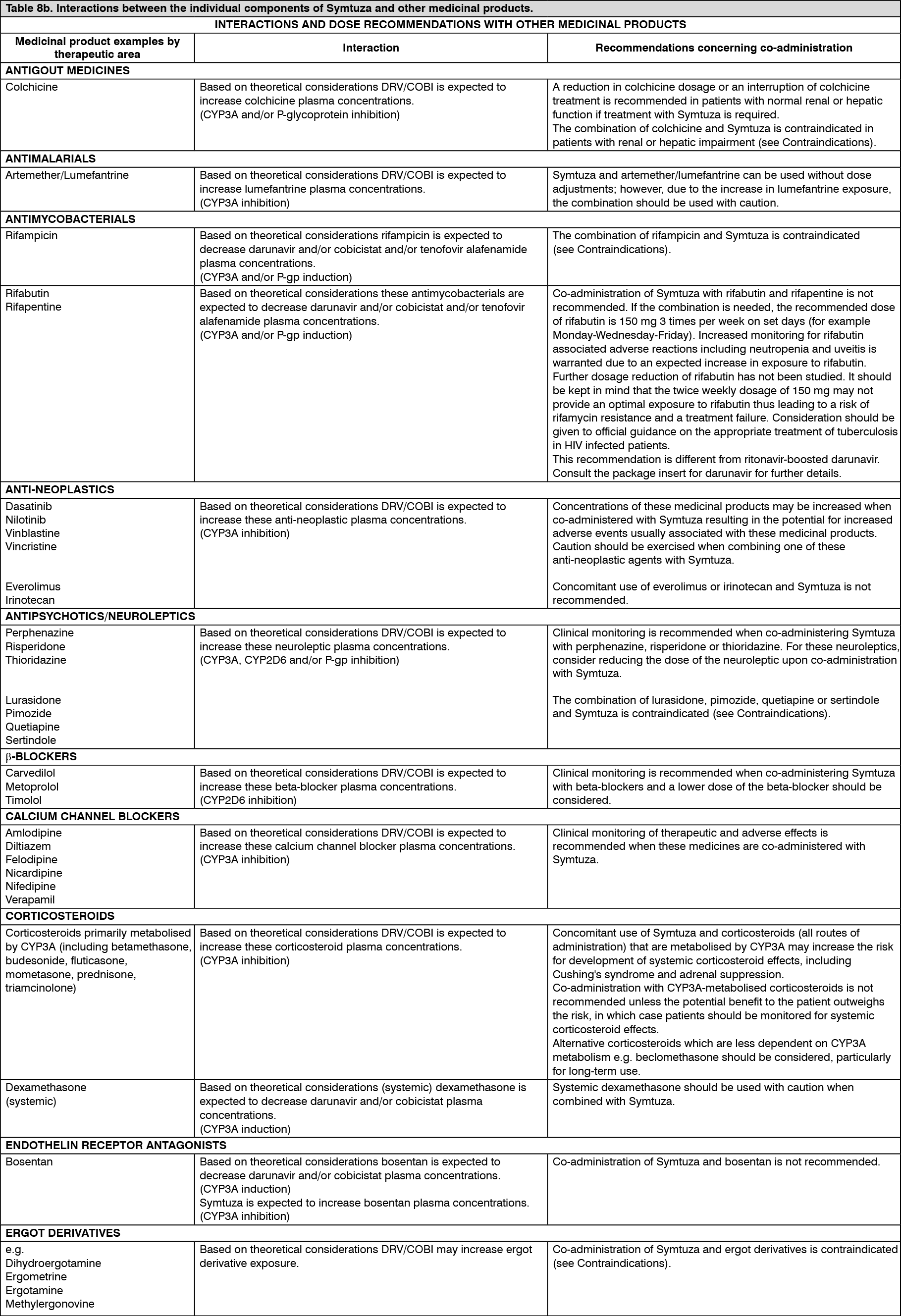 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image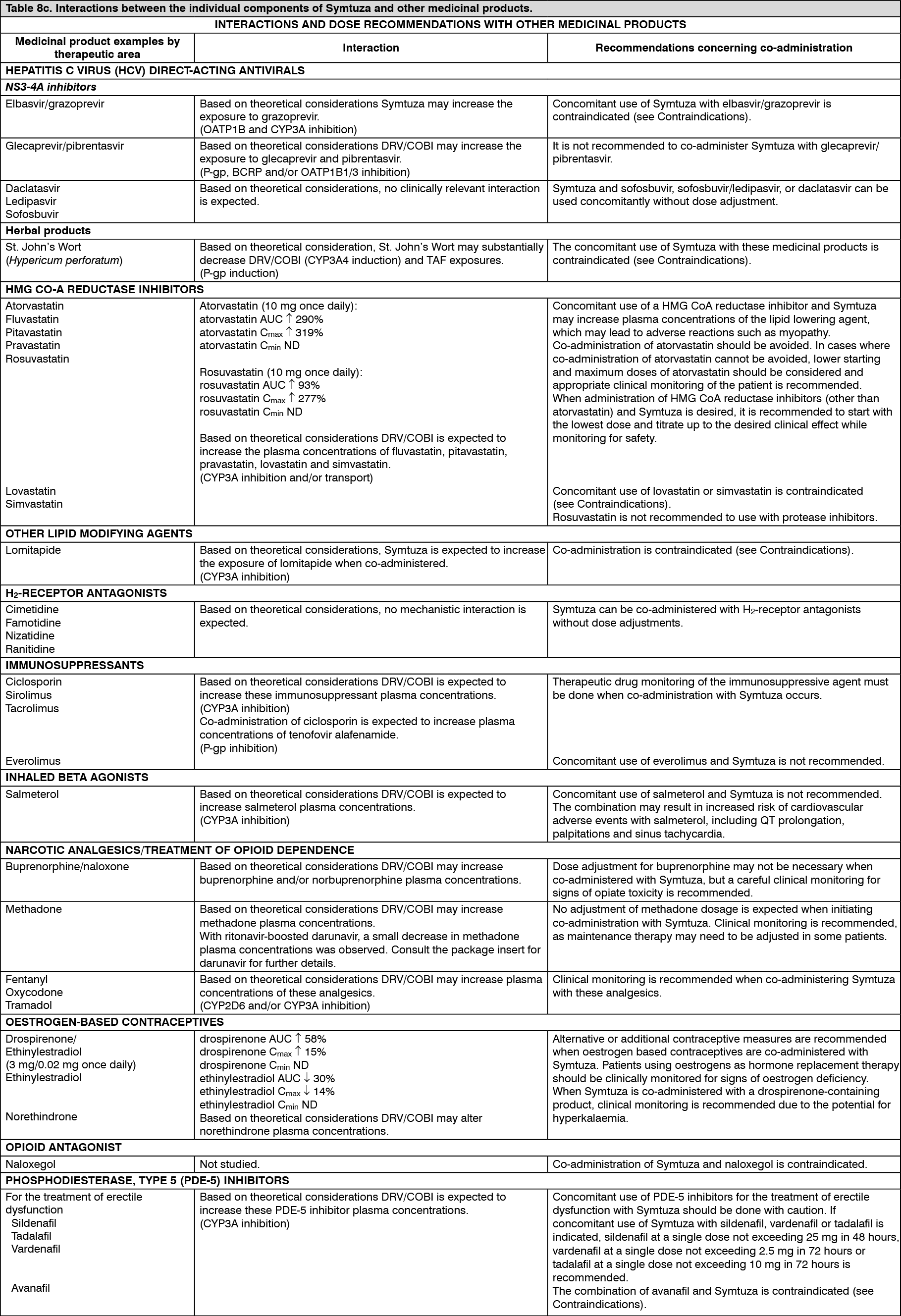 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image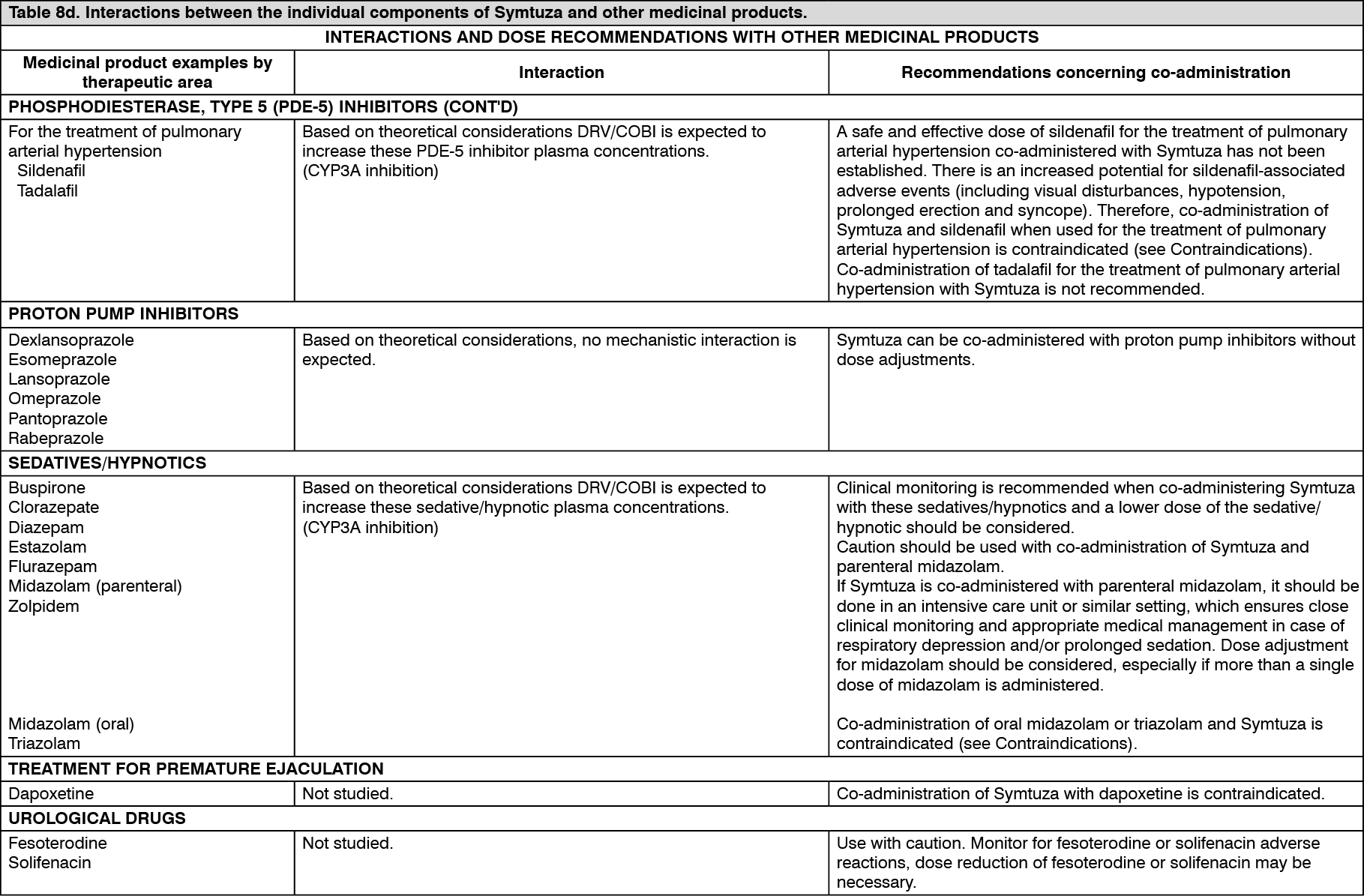 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out