Pharmacotherapeutic group: Antivirals for systemic use, other antivirals.
ATC code: J05AX12.
Pharmacology: Pharmacodynamics: Mechanism of action: Dolutegravir inhibits HIV integrase by binding to the integrase active site and blocking the strand transfer step of retroviral Deoxyribonucleic acid (DNA) integration which is essential for the HIV replication cycle.
Pharmacodynamic effects: Antiviral activity in cell culture: The IC
50 for dolutegravir in various labstrains using PBMC was 0.5 nM, and when using MT-4 cells it ranged from 0.7-2 nM. Similar IC50s were seen for clinical isolates without any major difference between subtypes; in a panel of 24 HIV-1 isolates of clades A, B, C, D, E, F and G and group O the mean IC
50 value was 0.2 nM (range 0.02-2.14). The mean IC
50 for 3 HIV-2 isolates was 0.18 nM (range 0.09-0.61).
Antiviral activity in combination with other antiviral agents: No antagonistic effects
in vitro were seen with dolutegravir and other antiretrovirals tested: stavudine, abacavir, efavirenz, nevirapine, lopinavir, amprenavir, enfuvirtide, maraviroc and raltegravir. In addition, no antagonistic effects were seen for dolutegravir and adefovir, and ribavirin had no apparent effect on dolutegravir activity.
Effect of human serum: In 100% human serum, the mean protein fold shift was 75 fold, resulting in protein adjusted IC
90 of 0.064 μg/mL.
Resistance: Resistance
in vitro: Serial passage is used to study resistance evolution
in vitro. When using the lab-strain HIV-1 IIIB during passage over 112 days, mutations selected appeared slowly, with substitutions at positions S153Y and F, resulting in a maximal fold change in susceptibility of 4 (range 2-4). These mutations were not selected in patients treated with dolutegravir in the clinical studies. Using strain NL432, mutations E92Q (FC 3) and G193E (also FC 3) were selected. The E92Q mutation has been selected in patients with pre-existing raltegravir resistance who were then treated with dolutegravir (listed as a secondary mutation for dolutegravir).
In further selection experiments using clinical isolates of subtype B, mutation R263K was seen in all five isolates (after 20 weeks and onwards). In subtype C (n=2) and A/G (n=2) isolates the integrase substitution R263K was selected in one isolate, and G118R in two isolates. R263K was reported from two ART experienced, INI naïve individual patients with subtypes B and C in the clinical program, but without effects on dolutegravir susceptibility
in vitro. G118R lowers the susceptibility to dolutegravir in site directed mutants (FC 10), but was not detected in patients receiving dolutegravir in the Phase III program.
Primary mutations for raltegravir/elvitegravir (Q148H/R/K, N155H, Y143R/H/C, E92Q and T66I) do not affect the
in vitro susceptibility of dolutegravir as single mutations. When mutations listed as secondary integrase inhibitor associated mutations (for raltegravir/elvitegravir) are added to these primary mutations in experiments with site directed mutants, dolutegravir susceptibility is still unchanged (FC <2 vs wild type virus), except in the case of Q148-mutations, where a FC of 5-10 or higher is seen with combinations of certain secondary mutations. The effect by the Q148-mutations (H/R/K) was also verified in passage experiments with site directed mutants. In serial passage with strain NL432, starting with site directed mutants harbouring N155H or E92Q, no further selection of resistance was seen (FC unchanged around 1). In contrast, starting with mutants harbouring mutation Q148H (FC 1), a variety of secondary mutations were seen with a consequent increase of FC to values >10.
A clinically relevant phenotypic cut-off value (FC vs wild type virus) has not been determined; genotypic resistance was a better predictor for outcome.
Seven hundred and five raltegravir resistant isolates from raltegravir experienced patients were analyzed for susceptibility to dolutegravir. Dolutegravir has a less than or equal to 10 FC against 94% of the 705 clinical isolates.
Resistance
in vivo: In previously untreated patients receiving dolutegravir + 2 NRTIs in Phase IIb and Phase III, no development of resistance to the integrase class, or to the NRTI class was seen (n=1118 follow-up of 48-96 weeks). In previously untreated patients receiving dolutegravir + lamivudine in the GEMINI studies through week 48 (n=716), no development of resistance to the integrase class, or to the NRTI class was seen.
In patients with prior failed therapies, but naïve to the integrase class (SAILING study), integrase inhibitor substitutions were observed in 4/354 patients (follow-up 48 weeks) treated with dolutegravir, which was given in combination with an investigator selected background regimen (BR). Of these four, two subjects had a unique R263K integrase substitution, with a maximum FC of 1.93, one subject had a polymorphic V151V/I integrase substitution, with maximum FC of 0.92, and one subject had pre-existing integrase mutations and is assumed to have been integrase experienced or infected with integrase resistant virus by transmission. The R263K mutation was also selected
in vitro (see as previously mentioned).
In the presence of integrase class-resistance (VIKING-3 study) the following mutations were selected in 32 patients with protocol defined virological failure (PDVF) through Week 24 and with paired genotypes (all treated with dolutegravir 50 mg twice daily + optimized background agents): L74L/M (n=1), E92Q (n=2), T97A (n=9), E138K/A/T (n=8), G140S (n=2), Y143H (n=1), S147G (n=1), Q148H/K/R (n=4), and N155H (n=1) and E157E/Q (n=1). Treatment emergent integrase resistance typically appeared in patients with a history of the Q148-mutation (baseline or historic). Five further subjects experienced PDVF between weeks 24 and 48, and 2 of these 5 had treatment emergent mutations. Treatment-emergent mutations or mixtures of mutations observed were L74I (n=1), N155H (n=2).
The VIKING-4 study examined dolutegravir (plus optimized background therapy) in subjects with primary genotypic resistance to INIs at Screening in 30 subjects. Treatment-emergent mutations observed were consistent with those observed in the VIKING-3 study.
In paediatric patients with prior failed therapies, but naïve to the integrase class, the integrase inhibitor substitution G118R was observed in 5/159 patients treated with dolutegravir, given in combination with an investigator selected background regimen. Of these five, 4 participants had additional integrase associated substitutions as follows: L74M, E138E/K, E92E/Q and T66I. Four of the 5 participants with emergent G118R had phenotypic data available. Dolutegravir FC (fold change as compared to wildtype virus) for these four participants ranged from 6 to 25-fold.
Effects on electrocardiogram: No relevant effects were seen on the QTc interval, with doses exceeding the clinical dose by approximately three fold.
Clinical efficacy and safety: Previously untreated patients: The efficacy of dolutegravir in HIV-infected, therapy naïve subjects is based on the analyses of 96-week data from two randomized, international, double-blind, active-controlled trials, SPRING-2 (ING113086) and SINGLE (ING114467). This is supported by 96 week data from an open-label, randomized and active-controlled study FLAMINGO (ING114915) and additional data from the open-label phase of SINGLE to 144 weeks. The efficacy of dolutegravir in combination with lamivudine in adults is supported by 96-week data from two identical 148-week, randomised, multicentre, double-blind, non-inferiority studies GEMINI-1 (204861) and GEMINI-2 (205543).
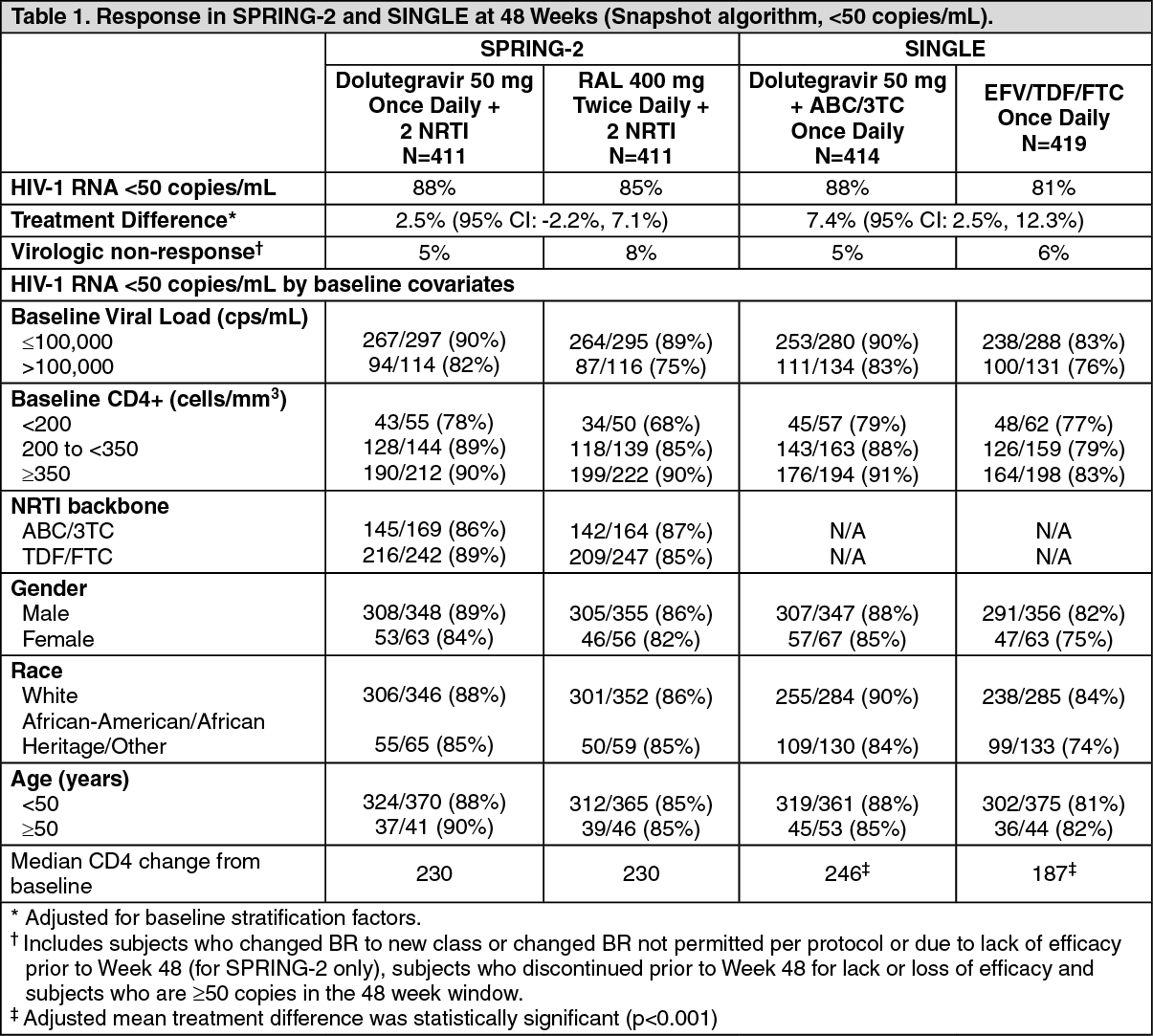
In SPRING-2, 822 adults were randomized and received at least one dose of either dolutegravir 50 mg once daily or raltegravir (RAL) 400 mg twice daily, both administered with either ABC/3TC or TDF/FTC. At baseline, median patient age was 36 years, 14% were female, 15% non-white, 11% had hepatitis B and/or C co-infection and 2% were CDC Class C, these characteristics were similar between treatment groups.
In SINGLE, 833 subjects were randomized and received at least one dose of either dolutegravir 50 mg once daily with fixed-dose abacavir-lamivudine (Dolutegravir + ABC/3TC) or fixed-dose efavirenz-tenofovir-emtricitabine (EFV/TDF/FTC). At baseline, median patient age was 35 years, 16% were female, 32% non-white, 7% had hepatitis C co-infection and 4% were CDC Class C, these characteristics were similar between treatment groups.
The primary endpoint and other week 48 outcomes (including outcomes by key baseline covariates) for SPRING-2 and SINGLE are shown in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
At week 48, dolutegravir was non-inferior to raltegravir in the SPRING-2 study, and in the SINGLE study dolutegravir + ABC/3TC was superior to efavirenz/TDF/FTC (p=0.003), Table 1. In SINGLE, the median time to viral suppression was shorter in the dolutegravir treated patients (28 vs 84 days, p<0.0001, analysis pre-specified and adjusted for multiplicity).
At week 96, results were consistent with those seen at week 48. In SPRING-2, dolutegravir was still non-inferior to raltegravir (viral suppression in 81% vs 76% of patients), and with a median change in CD4 count of 276 vs 264 cells/mm
3, respectively. In SINGLE, dolutegravir + ABC/3TC was still superior to EFV/TDF/FTC (viral suppression in 80% vs 72%), treatment difference 8.0% (2.3, 13.8), p=0.006, and with an adjusted mean change in CD4 count of 325 vs 281 cells/mm
3, respectively. At 144 weeks in the open-label phase of SINGLE, virologic suppression was maintained, the dolutegravir + ABC/3TC arm (71%) was superior to the EFV/TDF/FTC arm (63%), treatment difference was 8.3% (2.0, 14.6).
In FLAMINGO (ING114915), an open-label, randomised and active-controlled study, 484 HIV-1 infected antiretroviral naïve adults received one dose of either dolutegravir 50 mg once daily (n=242) or darunavir/ritonavir (DRV/r) 800 mg/100 mg once daily (n=242), both administered with either ABC/3TC or TDF/FTC. At baseline, median patient age was 34 years, 15% were female, 28% non-white, 10% had hepatitis B and/or C co-infection, and 3% were CDC Class C; these characteristics were similar between treatment groups. Virologic suppression (HIV-1 RNA <50 copies/mL) in the dolutegravir group (90%) was superior to the DRV/r group (83%) at 48 weeks. The adjusted difference in proportion and 95% CI were 7.1% (0.9, 13.2), p=0.025. At 96 weeks, virologic suppression in the dolutegravir group (80%) was superior to the DRV/r group (68%), adjusted treatment difference [Dolutegravir - (DRV+RTV)]: 12.4%; 95% CI: [4.7, 20.2].
In GEMINI-1 (204861) and GEMINI-2 (205543), identical 148-week, randomised, double-blind studies, 1433 adult HIV-1 infected antiretroviral naïve subjects were randomised to either a two-drug regimen of dolutegravir 50 mg plus lamivudine 300 mg once daily, or to a three-drug regimen of dolutegravir 50 mg once daily with fixed dose TDF/FTC. Subjects were enrolled with a screening plasma HIV-1 RNA of 1000 c/mL to ≤500,000 c/mL. At baseline, in the pooled analysis, median patient age was 33 years, 15% were female, 31% non-white, 6% had hepatitis C co-infection and 9% were CDC Stage 3. Approximately one third of the patients were infected with an HIV non-B subtype; these characteristics were similar between treatment groups. Virologic suppression (HIV-1 RNA <50 copies/mL) in the dolutegravir plus lamivudine group was non-inferior to the dolutegravir plus TDF/FTC group at 48 weeks, as shown in Table 2. The results of the pooled analysis were in line with those of the individual studies, for which the primary endpoint (difference in proportion <50 copies/mL plasma HIV-1 RNA at week 48 based on the Snapshot algorithm) was met. The adjusted difference was -2.6% (95% CI: -6.7; 1.5) for GEMINI-1 and -0.7% (95% CI: -4.3; 2.9) for GEMINI-2 with a prespecified non-inferiority margin of 10%. (See Table 2.)
Click on icon to see table/diagram/image
At 96 weeks the dolutegravir plus lamivudine group (86% with plasma HIV-1 RNA <50 copies/mL [pooled data]) remained non-inferior to the dolutegravir plus tenofovir/emtricitabine FDC group (90% with plasma HIV-1 RNA <50 copies/mL [pooled data]). The adjusted difference in proportions and 95% CI was -3.4% (-6.7, 0.0). The results of the pooled analysis were in line with those of the individual studies, for which the secondary endpoint (difference in proportion <50 copies/mL plasma HIV-1 RNA at Week 96 based on the Snapshot algorithm for dolutegravir plus lamivudine versus dolutegravir plus tenofovir/emtricitabine FDC) was met. The adjusted differences of -4.9 (95% CI: -9.8; 0.0) for GEMINI-1 and -1.8 (95% CI: -6.4; 2.7) for GEMINI-2 were within the prespecified non-inferiority margin of -10%. The mean increase in CD4+ T-cell counts was 269 in the DTG+3TC arm and 259 in the DTG+FTC/TDF arm, at week 96.
Treatment emergent resistance in previously untreated patients failing therapy: Through 96 weeks in SPRING-2 and FLAMINGO and 144 weeks in SINGLE, no cases of treatment emergent primary resistance to the integrase- or NRTI-class were seen in the dolutegravir-containing arms. For the comparator arms, the same lack of treatment emergent resistance was also the case for patients treated with darunavir/r in FLAMINGO. In SPRING-2, four patients in the RAL-arm failed with major NRTI mutations and one with raltegravir resistance; in SINGLE, six patients in the EFV/TDF/FTC-arm failed with mutations associated with NNRTI resistance, and one developed a major NRTI mutation. Through 96 weeks in the GEMINI-1 and GEMINI-2 studies, no cases of emergent resistance to the integrase- or NRTI-class were seen in either the Dolutegravir + 3TC or comparator Dolutegravir + TDF/FTC arms.
Patients with prior treatment failure, but not exposed to the integrase class: In the international multicentre, double-blind SAILING study (ING111762), 719 HIV-1 infected, antiretroviral therapy (ART)-experienced adults were randomized and received either dolutegravir 50 mg once daily or raltegravir 400 mg twice daily with investigator selected background regimen consisting of up to 2 agents (including at least one fully active agent). At baseline, median patient age was 43 years, 32% were female, 50% non-white, 16% had hepatitis B and/or C co-infection, and 46% were CDC Class C. All patients had at least two class ART resistance, and 49% of subjects had at least 3-class ART resistance at baseline.
Week 48 outcomes (including outcomes by key baseline covariates) for SAILING are shown in Table 3. (See Table 3.)
Click on icon to see table/diagram/image
In the SAILING study, virologic suppression (HIV-1 RNA <50 copies/mL) in the Tivicay arm (71%) was statistically superior to the raltegravir arm (64%), at Week 48 (p=0.03).
Statistically fewer subjects failed therapy with treatment-emergent integrase resistance on Tivicay (4/354, 1%) than on raltegravir (17/361, 5%) (p=0.003) (refer to Resistance: Resistance
in vivo as previously mentioned for details).
Patients with prior treatment failure that included an integrase inhibitor (and integrase class resistance): In the multicentre, open-label, single arm VIKING-3 study (ING112574), HIV-1 infected, ART-experienced adults with virological failure and current or historical evidence of raltegravir and/or elvitegravir resistance received Tivicay 50 mg twice daily with the current failing background regimen for 7 days but with optimised background ART from Day 8. The study enrolled 183 patients, 133 with INI-resistance at Screening and 50 with only historical evidence of resistance (and not at Screening). Raltegravir/elvitegravir was part of the current failing regimen in 98/183 patients (part of prior failing therapies in the others). At baseline, median patient age was 48 years, 23% were female, 29% non-white, and 20% had hepatitis B and/or C co-infection. Median baseline CD4+ was 140 cells/mm
3, median duration of prior ART was 14 years, and 56% were CDC Class C. Subjects showed multiple class ART resistance at baseline: 79% had ≥2 NRTI, 75% ≥1 NNRTI, and 71% ≥2 PI major mutations; 62% had non-R5 virus.
Mean change from baseline in HIV RNA at day 8 (primary endpoint) was -1.4log
10 copies/mL (95% CI -1.3 - -1.5log
10, p<0.001). Response was associated with baseline INI mutation pathway, as shown in Table 4. (See Table 4.)
Click on icon to see table/diagram/image
In patients without a primary mutation detected at baseline (N=60) (i.e. RAL/EVG not part of current failing therapy) there was a 1.63log
10 reduction in viral load at day 8.
After the functional monotherapy phase, subjects had the opportunity to re-optimize their background regimen when possible. The overall response rate through 24 weeks of therapy, 69% (126/183), was generally sustained through 48 weeks with 116/183 (63%) of patients with HIV-1 RNA <50 c/mL (ITT-E, Snapshot algorithm). When excluding patients who stopped therapy for non-efficacy reasons, and those with major protocol deviations (incorrect dolutegravir dosing, intake of prohibited co-medication), namely, "the Virological Outcome (VO)-population", the corresponding response rates were 75% (120/161, week 24) and 69% (111/160, week 48).
The response was lower when the Q148-mutation was present at baseline, and in particular in the presence of ≥2 secondary mutations, Table 5. The overall susceptibility score (OSS) of the optimised background regimen (OBR) was not associated with Week 24 response, nor with the week 48 response. (See Table 5.)
Click on icon to see table/diagram/image
The median change in CD4+ T cell count from baseline for VIKING-3 based on observed data was 61 cells/mm
3 at Week 24 and 110 cells/mm
3 at Week 48.
In the double blind, placebo controlled VIKING-4 study (ING116529), 30 HIV-1 infected, ART-experienced adults with primary genotypic resistance to INIs at Screening, were randomised to receive either dolutegravir 50 mg twice daily or placebo with the current failing regimen for 7 days followed by an open label phase with all subjects receiving dolutegravir. At baseline, median patient age was 49 years, 20% were female, 58% non-white, and 23% had hepatitis B and/or C co-infection. Median baseline CD4+ was 160 cells/mm
3, median duration of prior ART was 13 years, and 63% were CDC Class C. Subjects showed multiple class ART resistance at baseline: 80% had ≥2 NRTI, 73% ≥1 NNRTI, and 67% ≥2 PI major mutations; 83% had non-R5 virus. Sixteen of 30 subjects (53%) harboured Q148 virus at baseline. The primary endpoint at Day 8 showed that dolutegravir 50 mg twice daily was superior to placebo, with an adjusted mean treatment difference for the change from Baseline in Plasma HIV-1 RNA of -1.2log
10 copies/mL (95% CI -1.5 - -0.8log
10 copies/mL, p<0.001). The day 8 responses in this placebo controlled study were fully in line with those seen in VIKING-3 (not placebo controlled), including by baseline integrase resistance categories. At week 48, 12/30 (40%) subjects had HIV-1 RNA <50 copies/mL (ITT-E, Snapshot algorithm).
In a combined analysis of VIKING-3 and VIKING-4 (n=186, VO population), the proportion of subjects with HIV RNA <50 copies/mL at Week 48 was 123/186 (66%). The proportion of subjects with HIV RNA <50 copies/mL was 96/126 (76%) for No Q148 mutations, 22/41 (54%) for Q148+1 and 5/19 (26%) for Q148+≥2 secondary mutations.
Paediatric population: In an ongoing Phase I/II 48 week multicentre, open-label study (P1093/ING112578), the pharmacokinetic parameters, safety, tolerability and efficacy of dolutegravir film-coated tablets and dispersible tablets following once daily dosing were evaluated in combination regimens in HIV-1 infected infants, children and adolescents aged ≥4 weeks to <18 years, the majority of whom were treatment-experienced.
The efficacy results (Table 6) include participants who received the recommended once daily doses of either film-coated tablets or dispersible tablets. (See Table 6.)
Click on icon to see table/diagram/image
In participants experiencing virologic failure, 5/36 acquired integrase inhibitor substitution G118R. Of these five, 4 participants had additional integrase associated substitutions as follows: L74M, E138E/K, E92E/Q and T66I. Four of the 5 participants with emergent G118R had phenotypic data available. Dolutegravir FC (fold change as compared to wildtype virus) for these four participants ranged from 6 to 25-fold.
The European Medicines Agency has deferred the obligation to submit the results of studies with Tivicay in paediatric patients aged 4 weeks to below 6 years with HIV infection (see Dosage & Administration for information on paediatric use).
There are no data available on the use of dolutegravir plus lamivudine as a two-drug regimen in paediatric patients.
Pharmacokinetics: Dolutegravir pharmacokinetics are similar between healthy and HIV-infected subjects. The PK variability of dolutegravir is low to moderate. In Phase I studies in healthy subjects, between-subject CVb% for AUC and C
max ranged from ~20 to 40% and Cτ from 30 to 65% across studies. The between-subject PK variability of dolutegravir was higher in HIV-infected subjects than healthy subjects. Within-subject variability (CVw%) is lower than between-subject variability.
Absorption: Dolutegravir is rapidly absorbed following oral administration, with median T
max at 2 to 3 hours post dose for tablet formulation.
Food increased the extent and slowed the rate of absorption of dolutegravir. Bioavailability of dolutegravir depends on meal content: low, moderate, and high fat meals increased dolutegravir AUC
(0-∞) by 33%, 41%, and 66%, increased C
max by 46%, 52%, and 67%, prolonged T
max to 3, 4, and 5 hours from 2 hours under fasted conditions, respectively. These increases may be clinically relevant in the presence of certain integrase class resistance. Therefore, Tivicay is recommended to be taken with food by patients infected with HIV with integrase class resistance (see Dosage & Administration).
The absolute bioavailability of dolutegravir has not been established.
Distribution: Dolutegravir is highly bound (>99%) to human plasma proteins based on
in vitro data. The apparent volume of distribution is 17 L to 20 L in HIV-infected patients, based on a population pharmacokinetic analysis. Binding of dolutegravir to plasma proteins is independent of dolutegravir concentration. Total blood and plasma drug-related radioactivity concentration ratios averaged between 0.441 to 0.535, indicating minimal association of radioactivity with blood cellular components. The unbound fraction of dolutegravir in plasma is increased at low levels of serum albumin (<35 g/L) as seen in subjects with moderate hepatic impairment.
Dolutegravir is present in cerebrospinal fluid (CSF). In 13 treatment-naïve subjects on a stable dolutegravir plus abacavir/lamivudine regimen, dolutegravir concentration in CSF averaged 18 ng/mL (comparable to unbound plasma concentration, and above the IC
50).
Dolutegravir is present in the female and male genital tract. AUC in cervicovaginal fluid, cervical tissue and vaginal tissue were 6-10% of those in corresponding plasma at steady state. AUC in semen was 7% and 17% in rectal tissue of those in corresponding plasma at steady state.
Biotransformation: Dolutegravir is primarily metabolized through glucuronidation via UGT1A1 with a minor CYP3A component. Dolutegravir is the predominant circulating compound in plasma; renal elimination of unchanged active substance is low (<1% of the dose). Fifty-three percent of total oral dose is excreted unchanged in the faeces. It is unknown if all or part of this is due to unabsorbed active substance or biliary excretion of the glucuronidate conjugate, which can be further degraded to form the parent compound in the gut lumen. Thirty-two percent of the total oral dose is excreted in the urine, represented by ether glucuronide of dolutegravir (18.9% of total dose), N-dealkylation metabolite (3.6% of total dose), and a metabolite formed by oxidation at the benzylic carbon (3.0% of total dose).
Drug interactions: In vitro, dolutegravir demonstrated no direct, or weak inhibition (IC
50 >50 μM) of the enzymes cytochrome P450 (CYP)1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A, uridine diphosphate glucuronosyl transferase (UGT)1A1 or UGT2B7, or the transporters P-gp, BCRP, BSEP, OATP1B1, OATP1B3, OCT1, MATE2-K, MRP2 or MRP4.
In vitro, dolutegravir did not induce CYP1A2, CYP2B6 or CYP3A4. Based on this data, dolutegravir is not expected to affect the pharmacokinetics of medicinal products that are substrates of major enzymes or transporters (see Interactions).
In vitro, dolutegravir was not a substrate of human OATP1B1, OATP1B3 or OCT1.
Elimination: Dolutegravir has a terminal half-life of ~14 hours. The apparent oral clearance (CL/F) is approximately 1 L/hr in HIV-infected patients based on a population pharmacokinetic analysis.
Linearity/non-linearity: The linearity of dolutegravir pharmacokinetics is dependent on dose and formulation. Following oral administration of film-coated tablet formulations, in general, dolutegravir exhibited nonlinear pharmacokinetics with less than dose-proportional increases in plasma exposure from 2 to 100 mg; however increase in dolutegravir exposure appears dose proportional from 25 mg to 50 mg for the film-coated tablet formulation. With 50 mg film-coated tablet twice daily, the exposure over 24 hours was approximately doubled compared to 50 mg film-coated tablet once daily.
Pharmacokinetic/pharmacodynamic relationship(s): In a randomized, dose-ranging trial, HIV-1-infected subjects treated with dolutegravir monotherapy (ING111521) demonstrated rapid and dose-dependent antiviral activity, with mean decline in HIV-1 RNA of 2.5log
10 at day 11 for 50 mg dose. This antiviral response was maintained for 3 to 4 days after the last dose in the 50 mg group.
PK/PD modelling using pooled data from clinical studies in integrase resistant patients suggest that increasing the dose from 50 mg film-coated tablet twice daily to 100 mg film-coated tablet twice daily may increase the effectiveness of dolutegravir in patients with integrase resistance and limited treatment options due to advanced multi class resistance. The proportion of responders (HIV-1 RNA <50 c/mL) at week 24 was predicted to increase around 4-18% in the subjects with Q148+≥2 secondary mutations from G140A/C/S, E138A/K/T, L74I. Although these simulated results have not been confirmed in clinical trials, this high dose may be considered in the presence of the Q148+≥2 secondary mutations from G140A/C/S, E138A/K/T, L74I in patients with overall limited treatment options due to advanced multi class resistance. There is no clinical data on the safety or efficacy of the 100 mg film-coated tablet twice daily dose. Co-treatment with atazanavir increases the exposure of dolutegravir markedly, and should not be used in combination with this high dose, since safety with the resulting dolutegravir exposure has not been established.
Special patient populations: Children: The pharmacokinetics of dolutegravir given once daily as film-coated and dispersible tablets in HIV-1 infected infants, children and adolescents aged ≥4 weeks to <18 years were evaluated in two on-going studies (P1093/ING112578 and ODYSSEY/201296). Steady state simulated plasma exposure at once daily weight band doses is summarized in Table 7. (See Tables 7 and 8.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Elderly: Population pharmacokinetic analysis of dolutegravir using data in HIV-1 infected adults showed that there was no clinically relevant effect of age on dolutegravir exposure.
Pharmacokinetic data for dolutegravir in subjects >65 years of age are limited.
Renal impairment: Renal clearance of unchanged active substance is a minor pathway of elimination for dolutegravir. A study of the pharmacokinetics of a single 50 mg dose of dolutegravir film-coated tablets was performed in subjects with severe renal impairment (CLcr <30 mL/min) and matched healthy controls. The exposure to dolutegravir was decreased by approximately 40% in subjects with severe renal impairment. The mechanism for the decrease is unknown. No dosage adjustment is considered necessary for patients with renal impairment. Tivicay has not been studied in patients on dialysis.
Hepatic impairment: Dolutegravir is primarily metabolized and eliminated by the liver. A single 50 mg dose of dolutegravir film-coated tablets was administered to 8 subjects with moderate hepatic impairment (Child-Pugh class B) and to 8 matched healthy adult controls. While the total dolutegravir concentration in plasma was similar, a 1.5- to 2-fold increase in unbound exposure to dolutegravir was observed in subjects with moderate hepatic impairment compared to healthy controls. No dosage adjustment is considered necessary for patients with mild to moderate hepatic impairment. The effect of severe hepatic impairment on the pharmacokinetics of Tivicay has not been studied.
Polymorphisms in drug metabolising enzymes: There is no evidence that common polymorphisms in drug metabolising enzymes alter dolutegravir pharmacokinetics to a clinically meaningful extent. In a meta-analysis using pharmacogenomics samples collected in clinical studies in healthy subjects, subjects with UGT1A1 (n=7) genotypes conferring poor dolutegravir metabolism had a 32% lower clearance of dolutegravir and 46% higher AUC compared with subjects with genotypes associated with normal metabolism via UGT1A1 (n=41).
Gender: Population PK analyses using pooled pharmacokinetic data from Phase IIb and Phase III adult trials revealed no clinically relevant effect of gender on the exposure of dolutegravir.
Race: Population PK analyses using pooled pharmacokinetic data from Phase IIb and Phase III adult trials revealed no clinically relevant effect of race on the exposure of dolutegravir. The pharmacokinetics of dolutegravir following single dose oral administration to Japanese subjects appear similar to observed parameters in Western (US) subjects.
Co-infection with Hepatitis B or C: Population pharmacokinetic analysis indicated that hepatitis C virus co-infection had no clinically relevant effect on the exposure to dolutegravir. There are limited data on subjects with hepatitis B co-infection.
Toxicology: Preclinical safety data: Dolutegravir was not mutagenic or clastogenic using
in vitro tests in bacteria and cultured mammalian cells, and an
in vivo rodent micronucleus assay. Dolutegravir was not carcinogenic in long term studies in the mouse and rat.
Dolutegravir did not affect male or female fertility in rats at doses up to 1000 mg/kg/day, the highest dose tested (24 times the 50 mg twice daily human clinical exposure based on AUC).
Oral administration of dolutegravir to pregnant rats at doses up to 1000 mg/kg daily from days 6 to 17 of gestation did not elicit maternal toxicity, developmental toxicity or teratogenicity (27 times the 50 mg twice daily human clinical exposure based on AUC).
Oral administration of dolutegravir to pregnant rabbits at doses up to 1000 mg/kg daily from days 6 to 18 of gestation did not elicit developmental toxicity or teratogenicity (0.40 times the 50 mg twice daily human clinical exposure based on AUC). In rabbits, maternal toxicity (decreased food consumption, scant/no faeces/urine, suppressed body weight gain) was observed at 1000 mg/kg (0.40 times the 50 mg twice daily human clinical exposure based on AUC).
In a juvenile toxicity study in rats, dolutegravir administration resulted in two preweaning deaths at 75 mg/kg/day. Over the preweaning treatment period, mean body weight gain was decreased in this group and the decrease persisted throughout the entire study for females during the postweaning period. The systemic exposure at this dose (based on AUC) to dolutegravir was ~17-20-fold higher than humans at the recommended pediatric exposure. There were no new target organs identified in juveniles compared to adults. In the rat pre/post-natal development study, decreased body weight of the developing offspring was observed during lactation at a maternally toxic dose (approximately 27 times human exposure at the maximum recommended human dose).
The effect of prolonged daily treatment with high doses of dolutegravir has been evaluated in repeat oral dose toxicity studies in rats (up to 26 weeks) and in monkeys (up to 38 weeks). The primary effect of dolutegravir was gastrointestinal intolerance or irritation in rats and monkeys at doses that produce systemic exposures approximately 21 and 0.82 times the 50 mg twice daily human clinical exposure based on AUC, respectively. Because gastrointestinal (GI) intolerance is considered to be due to local active substance administration, mg/kg or mg/m
2 metrics are appropriate determinates of safety cover for this toxicity. GI intolerance in monkeys occurred at 15 times the human mg/kg equivalent dose (based on a 50 kg human), and 5 times the human mg/m
2 equivalent dose for a clinical dose of 50 mg twice daily.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image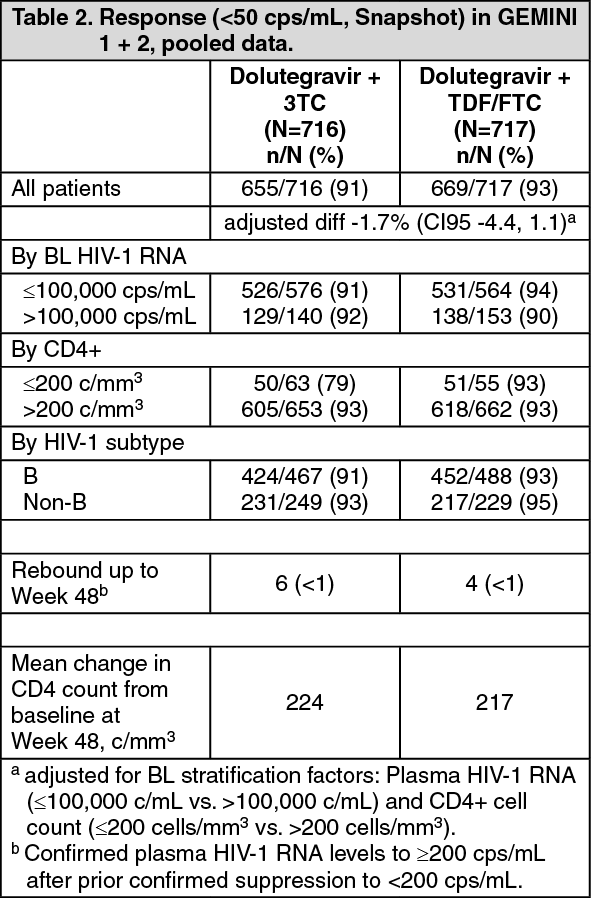 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image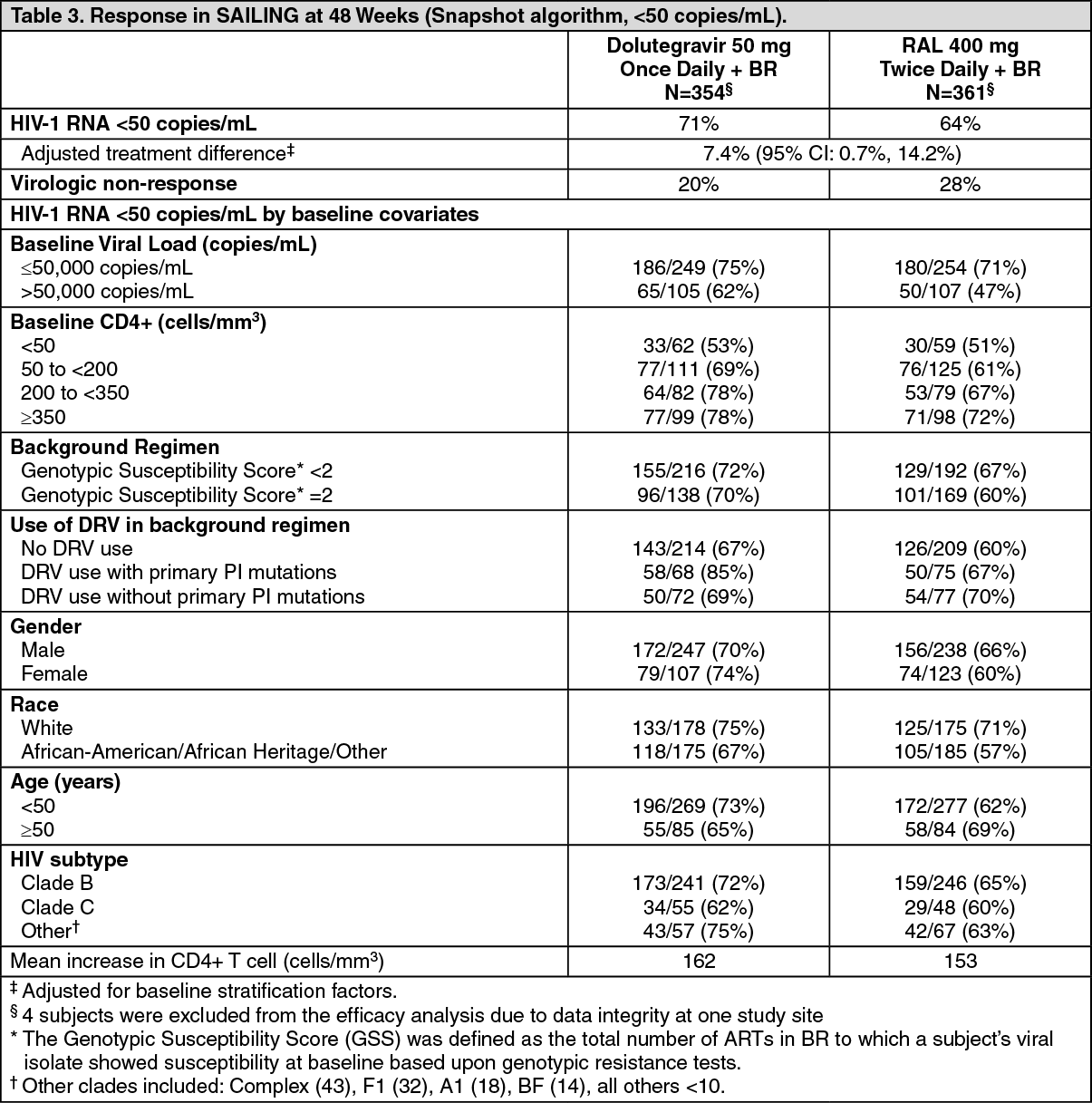 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image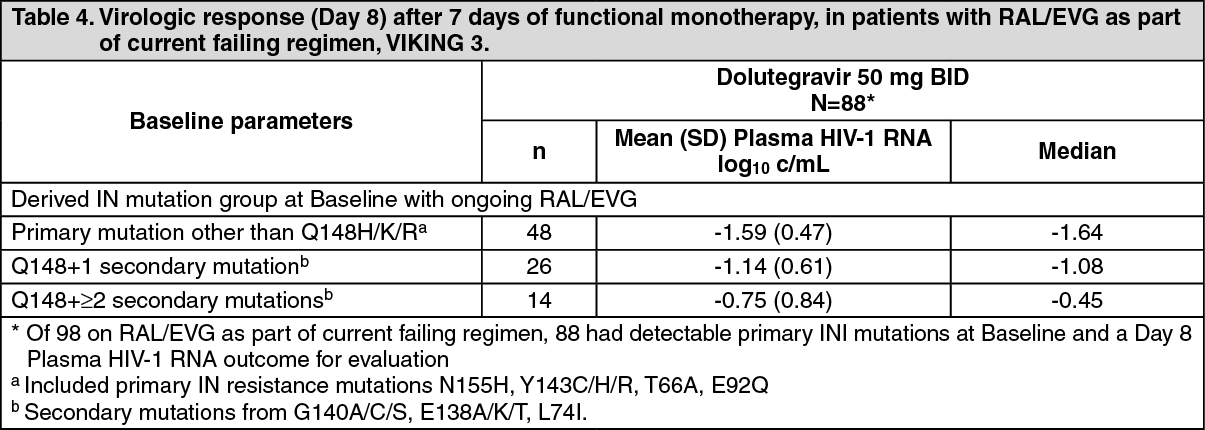 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image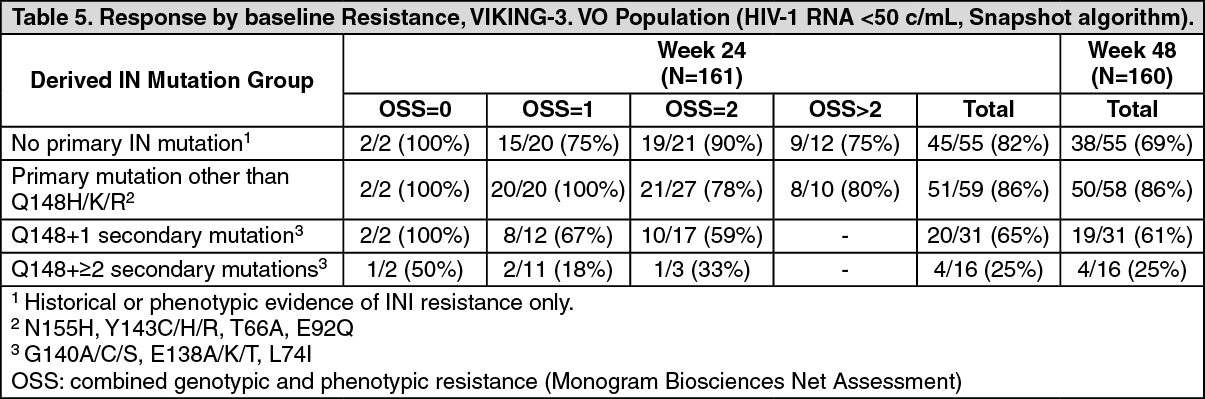 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image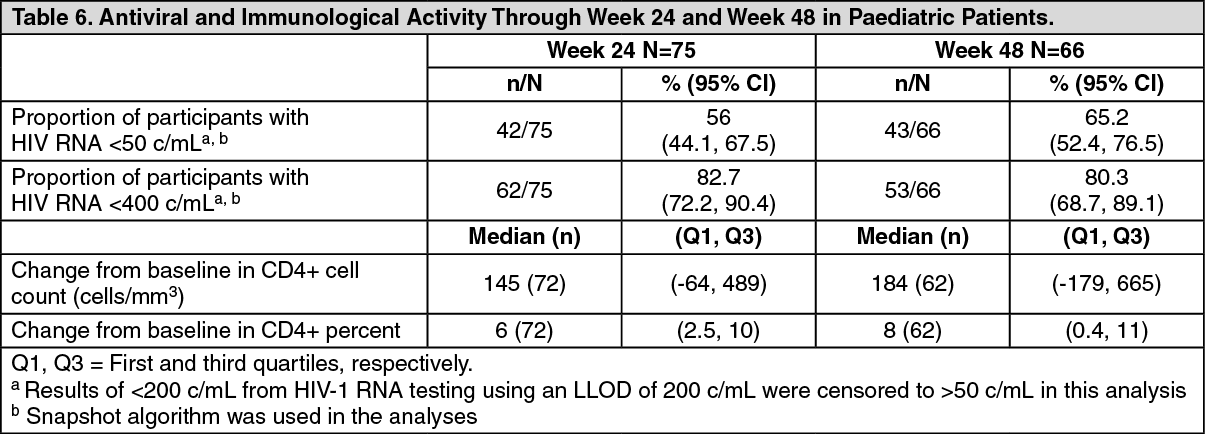 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image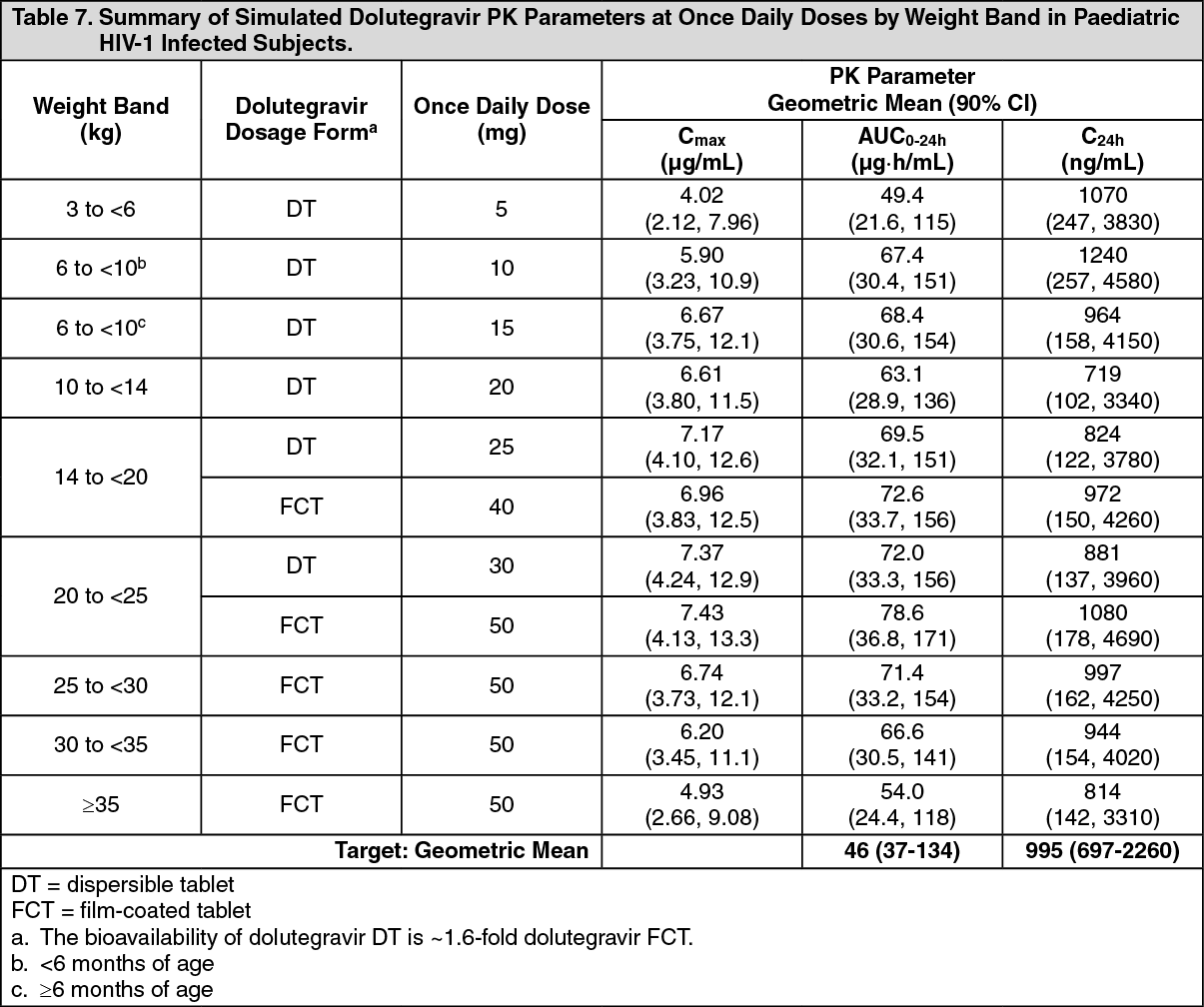 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image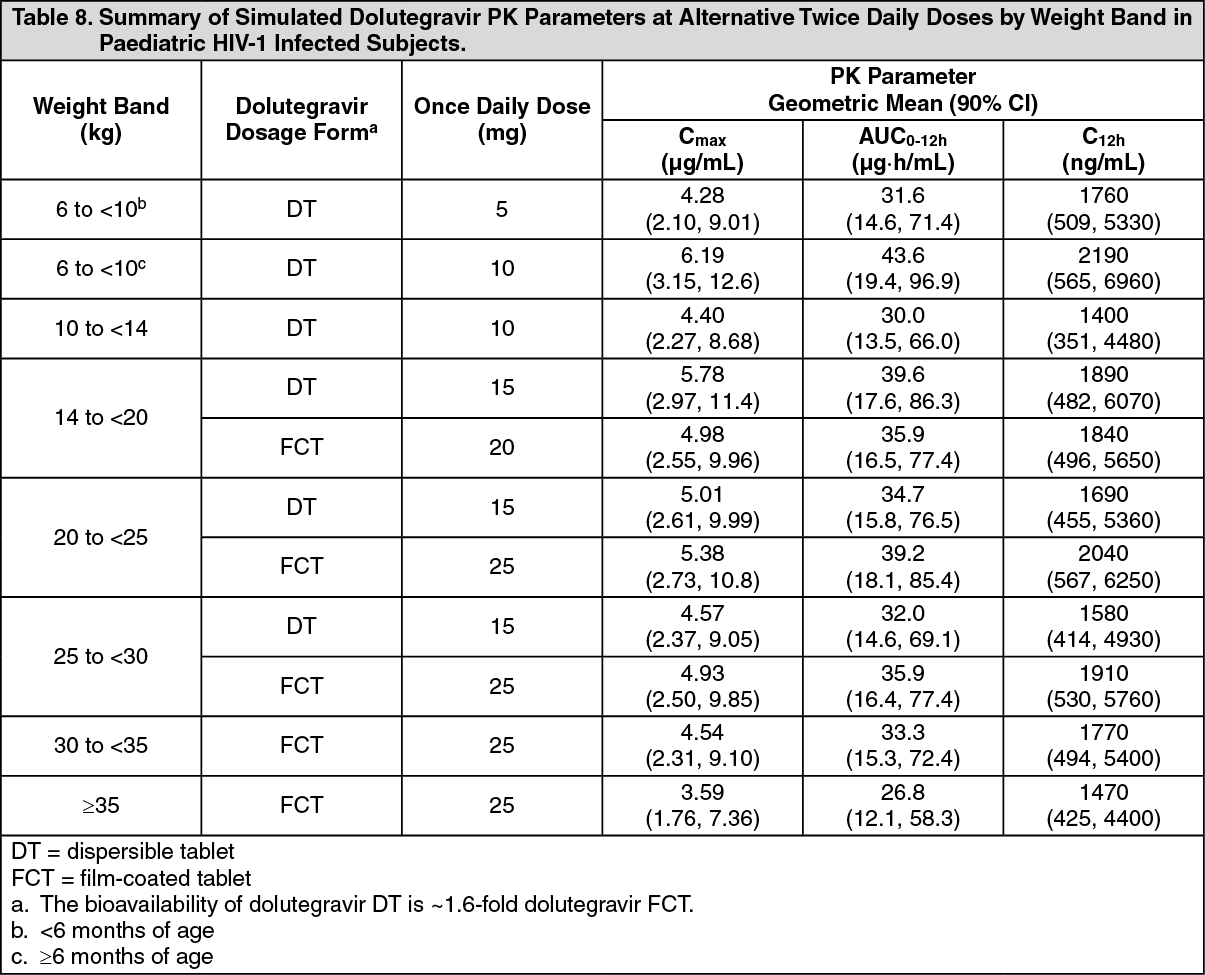 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
