Special precautions for disposal and other handling: Pump accessories found to be compatible with the administration of VELETRI include: CADD disposable Medication Cassette Reservoir 50 mL; 100 mL from Smiths Medical; CADD extension set with in-line 0.2 micron filter (CADD extension set with male luer, 0.2 micron air-eliminating filter, clamp, and integral anti-siphon valve with male luer) from Smiths Medical.
It is recommended that the infusion pump is not carried in permanent contact with the skin in order to avoid temperature excursions of the cassette.
Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
The stability of solutions of Veletri is pH dependent.
The powder for solution for infusion must be reconstituted using either Sterile Water for Injection or Sodium Chloride 0.9% Injection solution.
Further dilution should be performed with the same diluent as used for reconstitution of the sterile, lyophilised powder.
Reconstitution, dilution and calculation of infusion rate: Particular care should be taken in the preparation of the infusion and in calculating the rate of infusion. The following procedure given should be closely followed.
Reconstitution and dilution must be carried out under aseptic conditions.
Pulmonary Arterial Hypertension: There are two packs available for use in the treatment of pulmonary arterial hypertension, as follows: One vial containing sterile, freeze-dried Veletri equivalent to 0.5 mg Veletri supplied alone; One vial containing sterile, freeze-dried Veletri equivalent to 1.5 mg Veletri supplied alone.
Reconstitution: Withdraw 5 mL of either Sterile Water for Injection or Sodium Chloride 0.9% Injection diluent into a sterile syringe, inject the contents of the syringe into the vial containing Veletri and shake gently until the powder has dissolved. The reconstituted solution should be examined prior to further dilution. Its use is forbidden in the presence of discolouration or particles. Any unused reconstituted solution should be disposed of in accordance with local requirements.
Dilution: The reconstituted solution should be immediately further diluted to the final concentration.
Further dilution should be performed with the same diluent as used for reconstitution of the sterile, lyophilised powder.
Veletri when administered chronically, should be prepared in a drug delivery reservoir appropriate for the infusion pump. Only extension sets with an in-line 0.22 micron filter placed between the infusion pump and the catheter must be used. It is recommended to use filters with a hydrophilic polyethersulfone membrane. The extension set and the in-line filter must be changed at least every 48 hours (see Precautions).
The vial containing 0.5 mg epoprostenol must be used for the preparation of solutions with final concentrations below 15,000 ng/mL.
Table 2 provides examples for preparing frequently used concentrations of Veletri solutions. Each vial is for single use only. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Veletri diluted to the final concentration in the drug delivery reservoir as directed can be administered immediately at room temperature (25°C) or, if stored, for up to 8 days at 2 to 8°C as per the conditions of use outlined in Table 3. (See Table 3.)
Click on icon to see table/diagram/image
Do not expose the fully diluted solution to direct sunlight.
Calculation of infusion rate: Infusion rates may be calculated using the following formula: See equation.
Click on icon to see table/diagram/image
Examples for some concentrations commonly used in pulmonary arterial hypertension are shown as follows. (See Tables 4, 5 and 6.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Higher dosages, and therefore, more concentrated solutions may be necessary with long-term administration of Veletri.
Incompatibilities: This medicinal product must not be mixed with other medicinal products except those previously mentioned in Special precautions for disposal and other handling.


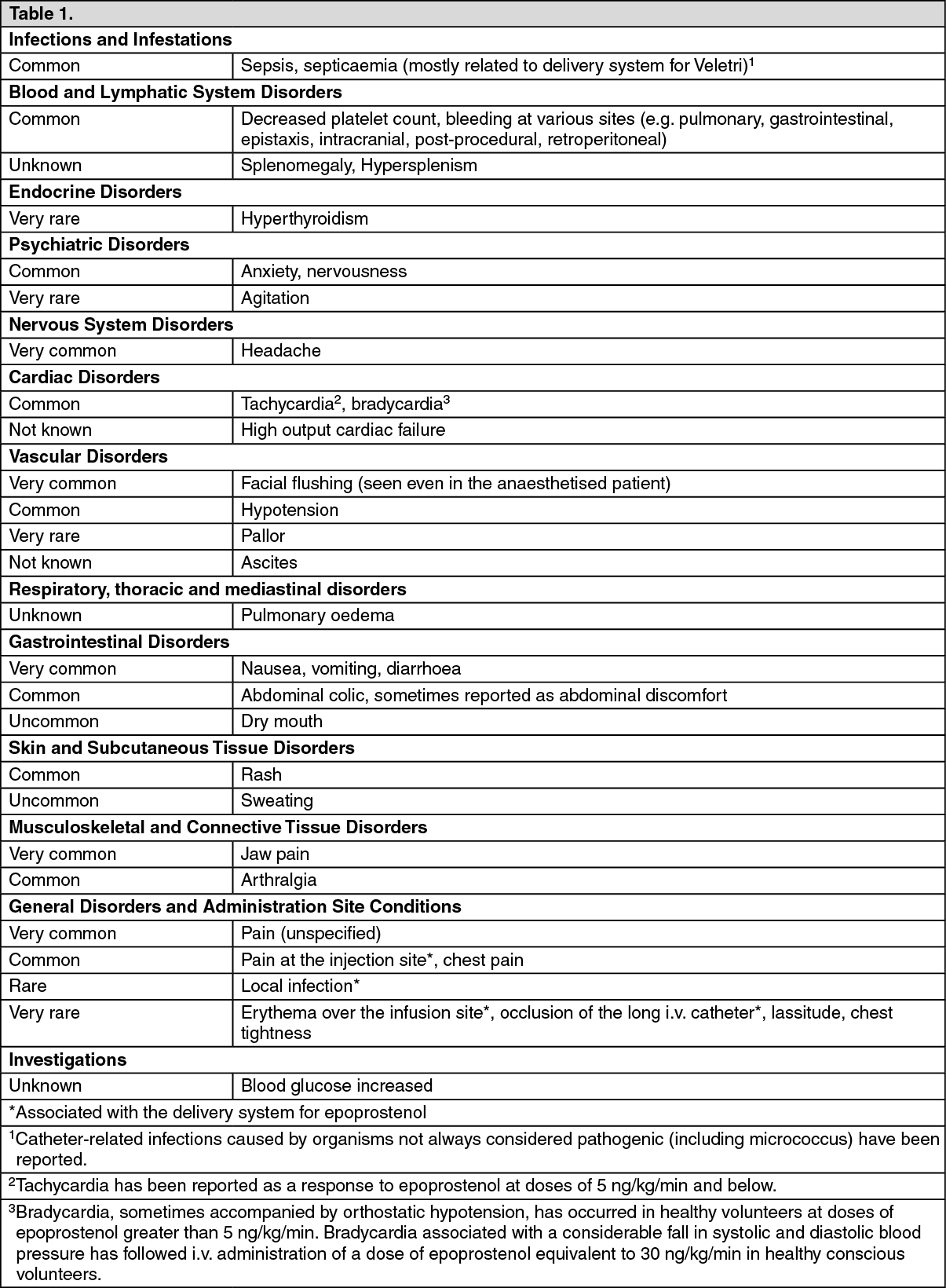 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image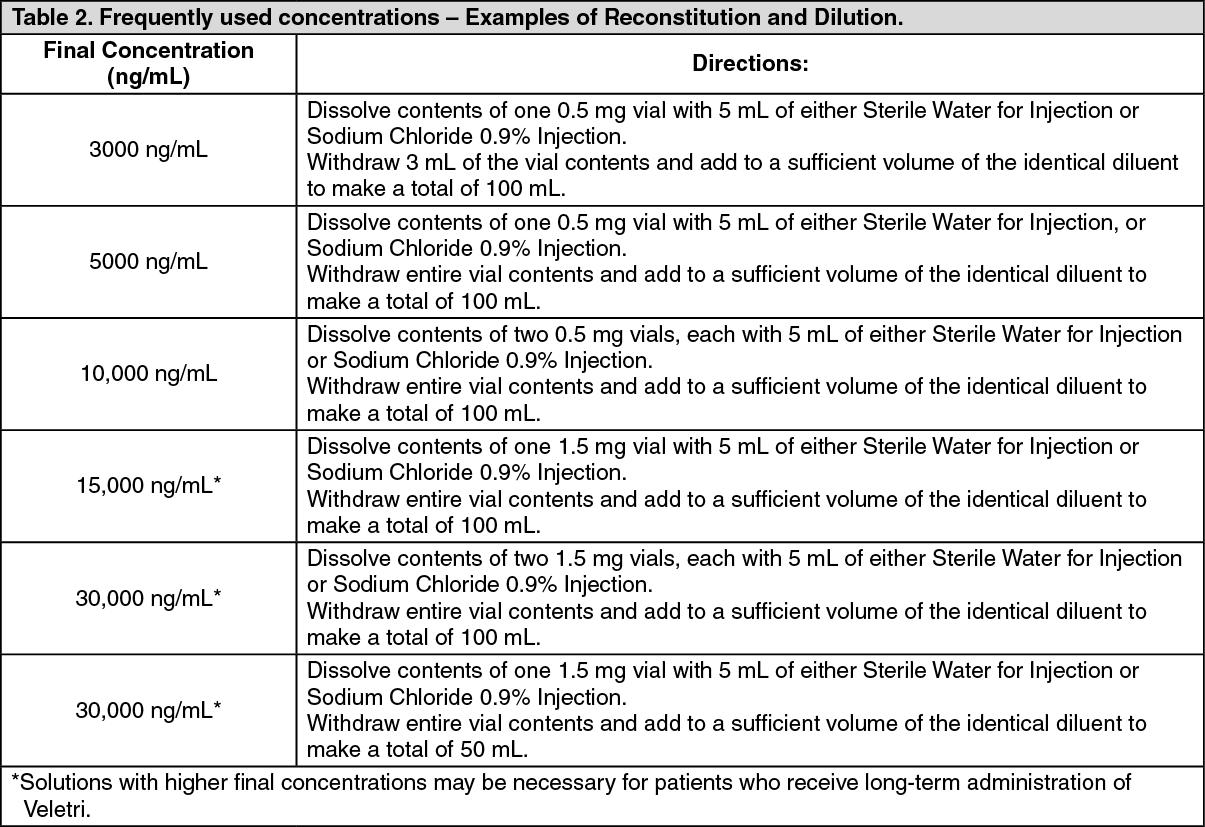 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image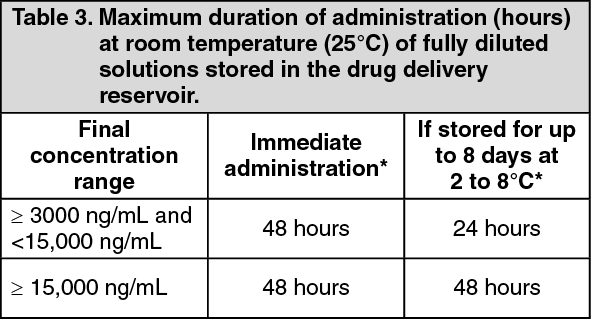 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image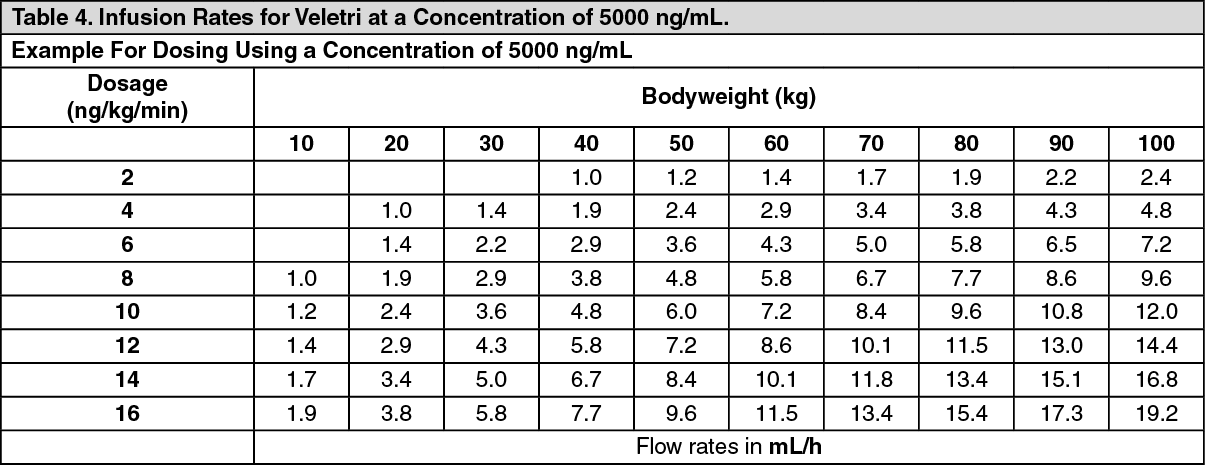 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image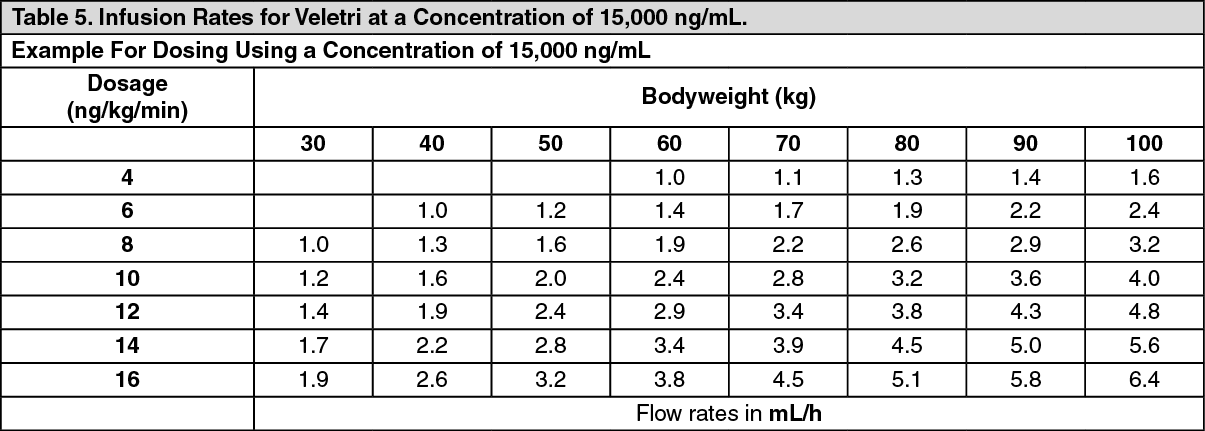 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image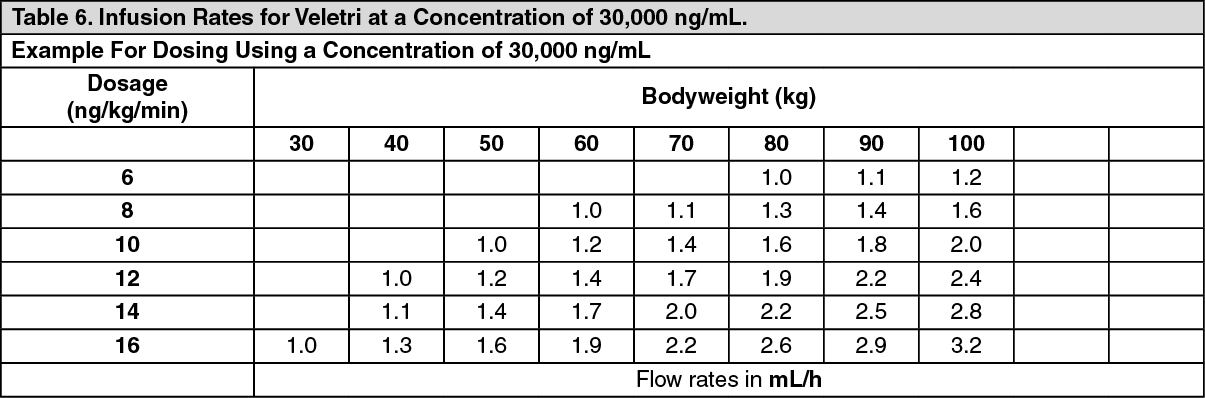 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out