


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Cabotegravir inhibits HIV integrase by binding to the integrase active site and blocking the strand transfer step of retroviral deoxyribonucleic acid (DNA) integration which is essential for the HIV replication cycle.
Pharmacodynamic effects: Antiviral activity in cell culture: Cabotegravir exhibited antiviral activity against laboratory strains of wild-type HIV-1 with mean concentration of cabotegravir necessary to reduce viral replication by 50 percent (EC50) values of 0.22 nM in peripheral blood mononuclear cells (PBMCs), 0.74 nM in 293T cells and 0.57 nM in MT-4 cells. Cabotegravir demonstrated antiviral activity in cell culture against a panel of 24 HIV-1 clinical isolates (three in each group of M clades A, B, C, D, E, F, and G, and 3 in group O) with EC50 values ranging from 0.02 nM to 1.06 nM for HIV-1. Cabotegravir EC50 values against three HIV-2 clinical isolates ranged from 0.10 nM to 0.14 nM. No clinical data is available in patients with HIV-2.
Antiviral activity in combination with other antiviral medicines/other medicinal products: No medicines with inherent anti-HIV activity were antagonistic to cabotegravir's antiretroviral activity (in vitro assessments were conducted in combination with rilpivirine, lamivudine, tenofovir and emtricitabine).
Resistance in vitro: Isolation from wild-type HIV-1 and activity against resistant strains: Viruses with >10-fold increase in cabotegravir EC50 were not observed during the 112-day passage of strain IIIB. The following integrase (IN) mutations emerged after passaging wild type HIV-1 (with T124A polymorphism) in the presence of cabotegravir: Q146L (fold-change [FC] range 1.3-4.6), S153Y [FC range 3.6-8.4 (Tablet)] [FC range 2.8-8.4 (Suspension for injection)], and I162M (FC = 2.8). As noted previously, the detection of T124A is selection of a pre-existing minority variant that does not have differential susceptibility to cabotegravir. No amino acid substitutions in the integrase region were selected when passaging the wild-type HIV-1 NL-432 in the presence of 6.4 nM of cabotegravir through Day 56.
Among the multiple mutants, the highest FC was observed with mutants containing Q148K or Q148R. E138K/Q148H resulted in a 0.92-fold decrease in susceptibility to cabotegravir but E138K/Q148R resulted in a 12-fold decrease in susceptibility and E138K/Q148K resulted in an 81-fold decrease in susceptibility to cabotegravir. G140C/Q148R and G140S/Q148R resulted in a 22- and 12-fold decrease in susceptibility to cabotegravir, respectively. While N155H did not alter susceptibility to cabotegravir, N155H/Q148R resulted in a 61-fold decrease in susceptibility to cabotegravir. Other multiple mutants, which resulted in a FC between 5 and 10, are: T66K/L74M (FC=6.3), G140S/Q148K (FC=5.6), G140S/Q148H (FC=6.1) and E92Q/N155H (FC=5.3).
Resistance in vivo: The number of subjects who met Confirmed Virologic Failure (CVF) criteria was low across the pooled FLAIR and ATLAS trials. In the pooled analysis, there were 7 CVFs on cabotegravir plus rilpivirine (7/591, 1.2%) and 7 CVFs on current antiretroviral regimen (7/591, 1.2%). The three CVFs on cabotegravir plus rilpivirine in FLAIR with resistance data had Subtype A1. In addition, 2 of the 3 CVFs had treatment-emergent integrase inhibitor resistance associated substitution Q148R while one of the three had G140R with reduced phenotypic susceptibility to cabotegravir. All 3 CVFs carried one rilpivirine resistance-associated substitution: K101E, E138E/A/K/T or E138K, and two of the three showed reduced phenotypic susceptibility to rilpivirine. The 3 CVFs in ATLAS had subtype A, A1 and AG. One of the three CVFs carried the INI resistance-associated substitution N155H at failure with reduced cabotegravir phenotype susceptibility. All three CVFs carried one rilpivirine resistance-associated substitution at failure: E138A, E138E/K or E138K, and showed reduced phenotypic susceptibility to rilpivirine. In two of these three CVFs, the rilpivirine resistance-associated substitutions observed at failure were also observed at baseline in PBMC HIV-1 DNA. The seventh CVF (FLAIR) never received an injection.
The substitutions associated with resistance to long-acting cabotegravir injection, observed in the pooled ATLAS and FLAIR trials were G140R (n=1), Q148R (n=2), and N155H (n=1).
In the ATLAS-2M study 10 subjects met CVF criteria through Week 48: 8 subjects (1.5%) in the Q8W arm and 2 subjects (0.4%) in the Q4W arm. Eight subjects met CVF criteria at or before the Week 24 timepoint.
At Baseline in the Q8W arm, 5 subjects had rilpivirine resistance-associated mutations of Y181Y/C + H221H/Y, Y188Y/F/H/L, Y188L, E138A or E138E/A and 1 subject contained cabotegravir resistance mutation, G140G/R (in addition to the previously mentioned Y188Y/F/H/L rilpivirine resistance-associated mutation). At the suspected virologic failure (SVF) timepoint in the Q8W arm, 6 subjects had rilpivirine resistance-associated mutations with 2 subjects having an addition of K101E and 1 subject having an addition of E138E/K from Baseline to SVF timepoint. Rilpivirine FC was above the clinical/biological cut-off for 7 subjects and ranged from 2.4 to 15. Five of the 6 subjects with rilpivirine resistance-associated substitution, also had INSTI resistance-associated substitutions, N155H (n=2); Q148R; Q148Q/R + N155N/H (n=2). INSTI substitution, L74I, was seen in 4/7 subjects. The Integrase genotype and phenotype assay failed for one subject and cabotegravir phenotype was unavailable for another. FCs for the Q8W subjects ranged from 0.6 to 9.1 for cabotegravir, 0.8 to 2.2 for dolutegravir and 0.8 to 1.7 for bictegravir.
In the Q4W arm, neither subject had any rilpivirine or INSTI resistance-associated substitutions at Baseline. One subject had the NNRTI substitution, G190Q, in combination with the NNRTI polymorphism, V189I. At SVF timepoint, one subject had on-treatment rilpivirine resistance-associated mutations, K101E + M230L and the other retained the G190Q + V189I NNRTI substitutions with the addition of V179V/I. Both subjects showed reduced phenotypic susceptibility to rilpivirine. Both subjects also had INSTI resistance-associated mutations, either Q148R + E138E/K or N155N/H at SVF and 1 subject had reduced susceptibility to cabotegravir. Neither subject had the INSTI substitution, L74I. FCs for the Q4W subjects were 1.8 and 4.6 for cabotegravir, 1.0 and 1.4 for dolutegravir and 1.1 and 1.5 for bictegravir.
Clinical efficacy and safety: The efficacy of Vocabria plus rilpivirine has been evaluated in two Phase III randomised, multicentre, active-controlled, parallel-arm, open-label, non-inferiority studies, FLAIR (study 201584) and ATLAS (study 201585). The primary analysis was conducted after all subjects completed their Week 48 visit or discontinued the study prematurely.
Patients virologically suppressed (on prior dolutegravir based regimen for 20 weeks): In FLAIR, 629 HIV-1-infected, antiretroviral treatment (ART)-naïve subjects received a dolutegravir integrase strand transfer inhibitor (INSTI) containing regimen for 20 weeks (either dolutegravir/abacavir/lamivudine or dolutegravir plus 2 other nucleoside reverse transcriptase inhibitors if subjects were HLA-B*5701 positive). Subjects who were virologically suppressed (HIV-1 RNA <50 copies per mL, n=566) were then randomised (1:1) to receive either the Vocabria plus rilpivirine regimen or remain on the current antiretroviral (CAR) regimen. Subjects randomised to receive the Vocabria plus rilpivirine regimen, initiated treatment with oral lead-in dosing with one 30 mg Vocabria tablet plus one 25 mg rilpivirine tablet, daily, for at least 4 weeks, followed by treatment with Vocabria injection (month 1: 600 mg injection, month 2 onwards: 400 mg injection) plus rilpivirine injection (month 1: 900 mg injection, month 2 onwards: 600 mg injection) every month for an additional 44 weeks. This study was extended to 96 weeks.
Patients virologically suppressed (stable on prior ARV therapy for at least 6 months): In ATLAS, 616 HIV-1-infected, ART-experienced, virologically-suppressed (for at least 6 months) subjects (HIV-1 RNA <50 copies per mL) were randomised (1:1) and received either the Vocabria plus rilpivirine regimen or remained on the CAR regimen. Subjects randomised to receive the Vocabria plus rilpivirine regimen, initiated treatment with oral lead-in dosing with one 30 mg Vocabria tablet plus one 25 mg rilpivirine tablet, daily for at least 4 weeks, followed by treatment with Vocabria injection (month 1: 600 mg injection, month 2 onwards: 400 mg injection) plus rilpivirine injection (month 1: 900 mg injection, month 2 onwards: 600 mg injection) every month for an additional 44 weeks. In ATLAS, 50%, 17%, and 33% of subjects received an NNRTI, PI, or INI (respectively) as their baseline third treatment medicine class prior to randomisation and this was similar between treatment arms.
Pooled data: At baseline, in the pooled analysis, for the Vocabria plus rilpivirine arm, the median age of subjects was 38 years, 27% were female, 27% were non-white, 1% were ≥65 years and 7% had CD4+ cell count less than 350 cells per mm3; these characteristics were similar between treatment arms.
The primary endpoint of both studies was the proportion of subjects with plasma HIV-1 RNA ≥50 copies/mL at week 48 (snapshot algorithm for the ITT-E population).
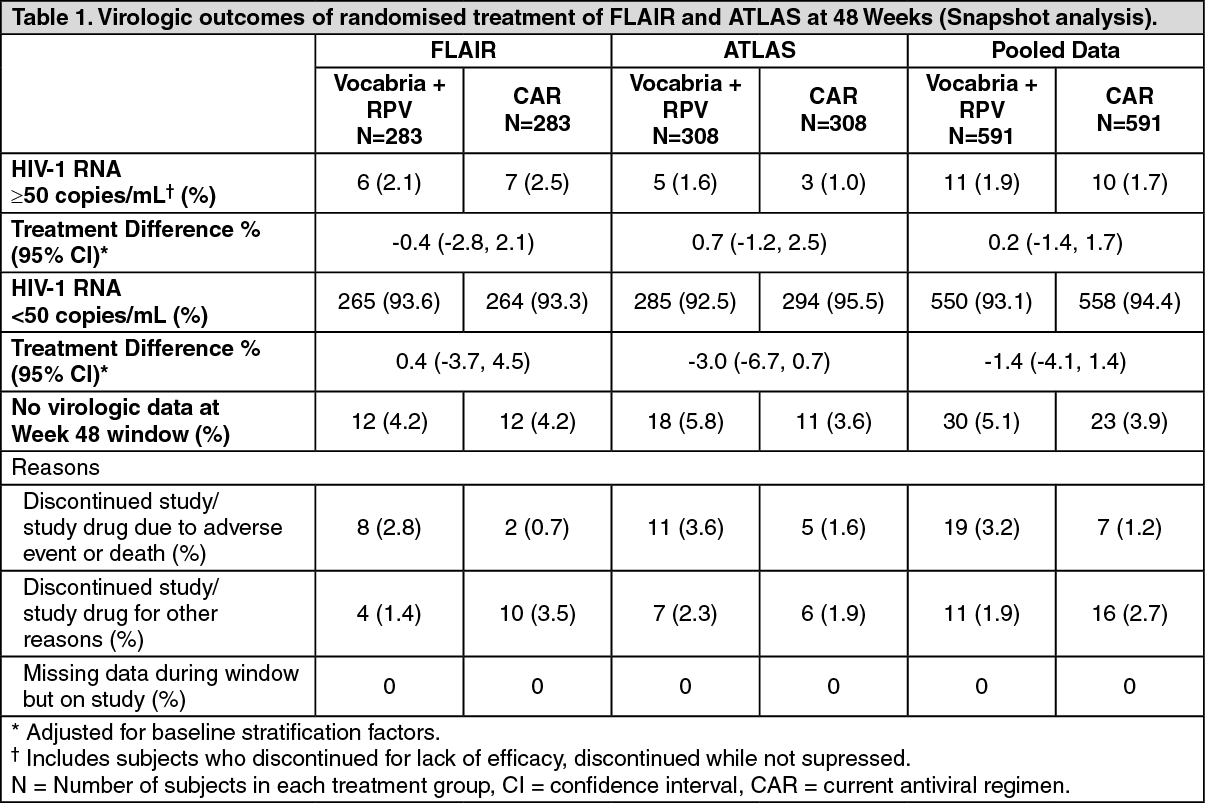
In a pooled analysis of the two pivotal studies, Vocabria plus rilpivirine was non-inferior to CAR on the proportion of subjects having plasma HIV-1 RNA ≥50 c/mL (1.9% and 1.7% respectively) at Week 48. The adjusted treatment difference between Vocabria plus rilpivirine and CAR (0.2; 95% CI: -1.4, 1.7) for the pooled analysis met the non-inferiority criterion (upper bound of the 95% CI below 4%).
The primary endpoint and other week 48 outcomes, including outcomes by key baseline factors, for FLAIR and ATLAS are shown in Tables 1 and 2. (See Tables 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image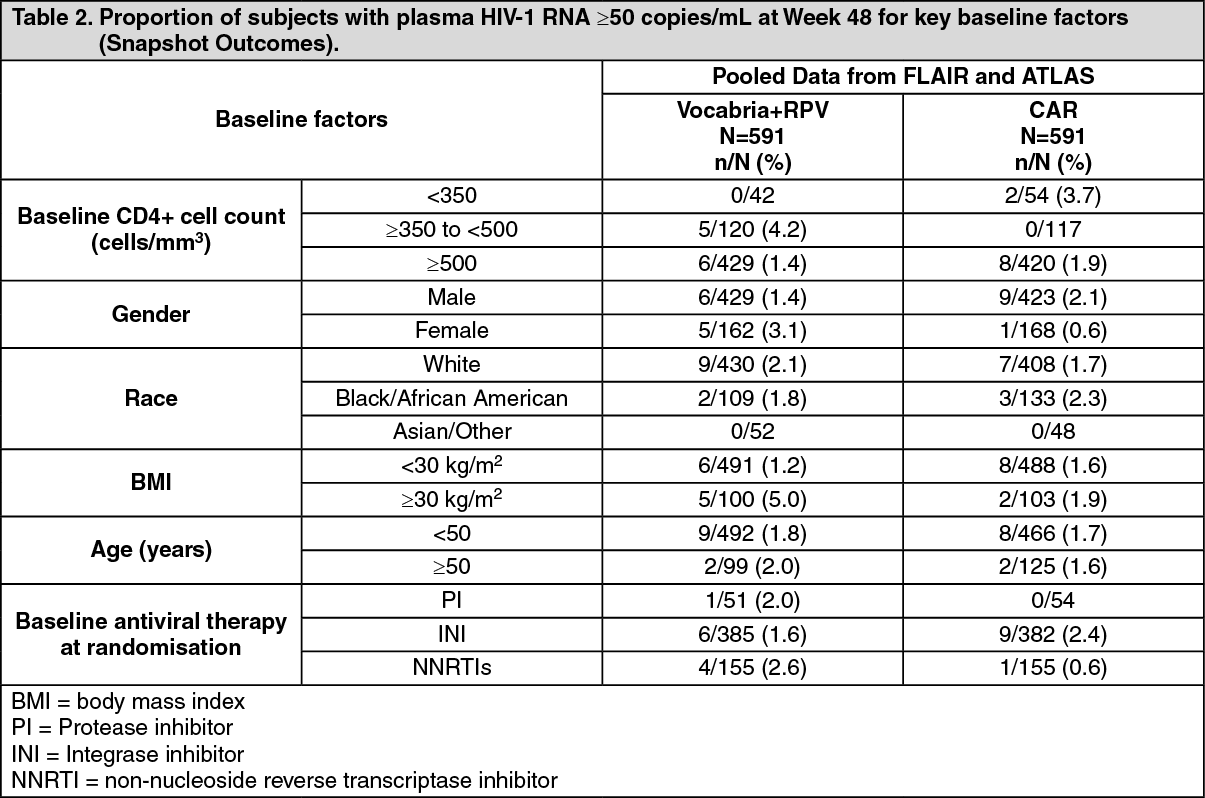 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn both the FLAIR and ATLAS studies, treatment differences across baseline characteristics (CD4+ count, gender, race, BMI, age, baseline third agent/medicine treatment class) were comparable.
Week 96 FLAIR: In the FLAIR study at 96 Weeks, the results remained consistent with the results at 48 Weeks. The proportion of subjects having plasma HIV-1 RNA ≥50 c/mL in Vocabria plus rilpivirine (n=283) and CAR (n=283) was 3.2% and 3.2% respectively (adjusted treatment difference between Vocabria plus rilpivirine and CAR [0.0; 95% CI: -2.9, 2.9]). The proportion of subjects having plasma HIV-1 RNA <50 c/mL in Vocabria plus rilpivirine and CAR was 87% and 89%, respectively (adjusted treatment difference between Vocabria plus rilpivirine and CAR [-2.8; 95% CI: -8.2, 2.5]).
Suspension for injection: Week 124 FLAIR Direct to Injection vs Oral Lead-in: In the FLAIR study, an evaluation of safety and efficacy was performed at Week 124 for patients electing to switch (at Week 100) from abacavir/dolutegravir/lamivudine to Vocabria plus rilpivirine in the Extension Phase. Subjects were given the option to switch with or without an oral lead-in phase, creating an oral lead-in (OLI) group (n=121) and a direct to injection (DTI) group (n=111).
At Week 124, the proportion of subjects with HIV-1 RNA ≥50 copies/mL was 0.8% and 0.9% for the oral lead-in and direct to injection groups, respectively. The rates of virologic suppression (HIV-1 RNA <50 c/mL) were similar in both OLI (93.4%) and DTI (99.1%) groups.
Every 2 month dosing: Patients virologically suppressed (stable on prior ARV therapy for at least 6 months): The efficacy and safety of Vocabria injection given every 2 months, has been evaluated in one Phase IIIb randomised, multicentre, parallel-arm, open-label, non-inferiority study, ATLAS-2M (207966). The primary analysis was conducted after all subjects completed their Week 48 visit or discontinued the study prematurely.
In ATLAS-2M, 1045 HIV-1 infected, ART experienced, virologically suppressed subjects were randomised (1:1) and received a Vocabria plus rilpivirine injection regimen administered either every 2 months or monthly. Subjects initially on non-cabotegravir/rilpivirine treatment received oral lead-in treatment comprising one 30 mg Vocabria tablet plus one 25 mg rilpivirine tablet, daily, for at least 4 weeks. Subjects randomised to monthly Vocabria injections (month 1: 600 mg injection, month 2 onwards: 400 mg injection) and rilpivirine injections (month 1: 900 mg injection, month 2 onwards: 600 mg injection) received treatment for an additional 44 weeks. Subjects randomised to every 2 month Vocabria injections (600 mg injection at months 1, 2, 4 and every 2 months thereafter) and rilpivirine injections (900 mg injection at months 1, 2, 4 and every 2 months thereafter) received treatment for an additional 44 weeks. Prior to randomisation, 63%, 13% and 24% of subjects received Vocabria plus rilpivirine for 0 weeks, 1 to 24 weeks and >24 weeks, respectively.
At baseline, the median age of subjects was 42 years, 27% were female, 27% were non-white, 4% were ≥65 years and 6% had a CD4+ cell count less than 350 cells per mm3; these characteristics were similar between the treatment arms.
The primary endpoint in ATLAS-2M was the proportion of subjects with a plasma HIV-1 RNA ≥50 c/mL at Week 48 (snapshot algorithm for the ITT-E population).
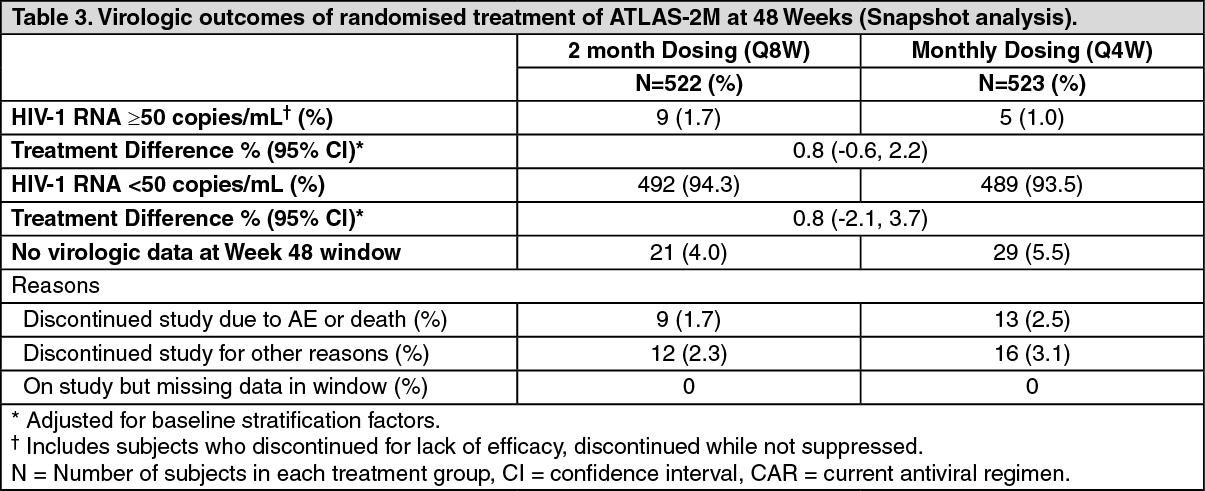
In ATLAS-2M, Vocabria and rilpivirine administered every 2 months was non-inferior to Vocabria and rilpivirine administered every month on the proportion of subjects having plasma HIV-1 RNA ≥50 c/mL (1.7% and 1.0% respectively) at Week 48. The adjusted treatment difference between Vocabria and rilpivirine administered every 2 months and every month (0.8; 95% CI: -0.6, 2.2) met the non-inferiority criterion (upper bound of the 95% CI below 4%). (See Tables 3 and 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image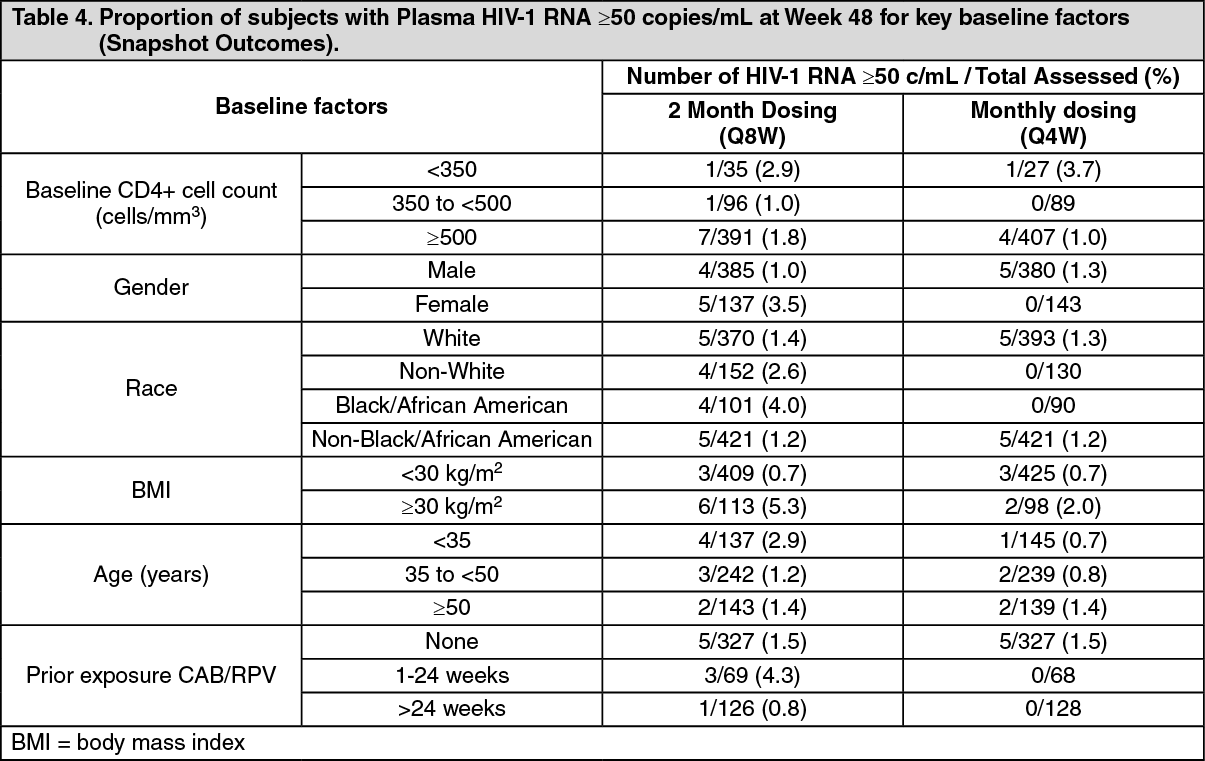 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the ATLAS-2M study, treatment differences on the primary endpoint across baseline characteristics (CD4+ lymphocyte count, gender, race, BMI, age and prior exposure to cabotegravir/rilpivirine) were not clinically meaningful.
Suspension for injection: The efficacy results at Week 96 are consistent with the results of the primary endpoint at Week 48. Vocabria plus rilpivirine injections administered every 2 months is non-inferior to Vocabria and rilpivirine administered every month. The proportion of subjects having plasma HIV-1 RNA ≥50 c/mL at Week 96 in Vocabria plus rilpivirine every 2 months dosing (n=522) and Vocabria plus rilpivirine monthly dosing (n=523) was 2.1% and 1.1% respectively (adjusted treatment difference between Vocabria plus rilpivirine every 2 months dosing and monthly dosing [1.0; 95% CI: -0.6, 2.5]). The proportion of subjects having plasma HIV-1 RNA <50 c/mL at Week 96 in Vocabria plus rilpivirine every 2 months dosing and Vocabria plus rilpivirine monthly dosing was 91% and 90.2% respectively (adjusted treatment difference between Vocabria plus rilpivirine every 2 months dosing and monthly dosing [0.8; 95% CI: -2.8, 4.3]).
The efficacy results at Week 152 are consistent with the results of the primary endpoint at Week 48 and at Week 96. Vocabria plus rilpivirine injections administered every 2 months is non-inferior to Vocabria and rilpivirine administered every month. In an ITT analysis, the proportion of subjects having plasma HIV-1 RNA ≥50 c/mL at Week 152 in Vocabria plus rilpivirine every 2 months dosing (n=522) and Vocabria plus rilpivirine monthly dosing (n=523) was 2.7% and 1.0% respectively (adjusted treatment difference between Vocabria plus rilpivirine every 2 months dosing and monthly dosing [1.7; 95% CI: 0.1, 3.3]). In an ITT analysis, the proportion of subjects having plasma HIV-1 RNA <50 c/mL at Week 152 in Vocabria plus rilpivirine every 2 months dosing and Vocabria plus rilpivirine monthly dosing was 87% and 86% respectively (adjusted treatment difference between Vocabria plus rilpivirine every 2 months dosing and monthly dosing [1.5; 95% CI: -2.6, 5.6]).
Post-hoc analyses: Tablet: Multivariable analyses of pooled phase 3 studies (ATLAS, FLAIR and ATLAS-2M), including data from 1039 HIV-infected adults with no prior exposure to Vocabria plus rilpivirine, examined the influence of baseline viral and participant characteristics, dosing regimen, and post-baseline plasma drug concentrations on confirmed virologic failure (CVF) using regression modelling with a variable selection procedure. Through Week 48 in these studies, 13/1039 (1.25%) participants had CVF while receiving cabotegravir and rilpivirine.
Four covariates were significantly associated (P<0.05 for each adjusted odds ratio) with increased risk of CVF: rilpivirine resistance mutations at baseline identified by proviral DNA genotypic assay, HIV-1 subtype A6/A1 (associated with integrase L74I polymorphism), rilpivirine trough concentration 4 weeks following initial injection dose, body mass index of at least 30 kg/m2 (associated with cabotegravir pharmacokinetics). Other variables including Q4W or Q8W dosing, female gender, or other viral subtypes (non A6/A1) had no significant association with CVF. No baseline factor, when present in isolation, was predictive of virologic failure. However, a combination of at least 2 of the following baseline factors was associated with an increased risk of CVF: rilpivirine resistance mutations, HIV-1 subtype A6/A1, or BMI ≥30 kg/m2 (see Table 5).
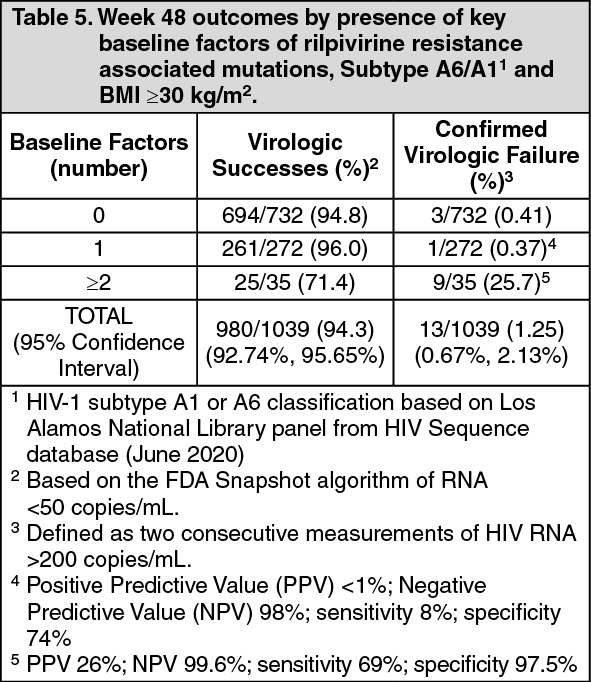 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSuspension for injection: Multivariable analyses of pooled phase 3 studies (ATLAS through 96 weeks, FLAIR through 124 weeks and ATLAS-2M through 152 weeks) examined the influence of various factors on the risk of CVF. The baseline factors analysis (BFA) examined baseline viral and participant characteristics and dosing regimen; and the multivariable analysis (MVA) included the baseline factors and incorporated post-baseline predicted plasma drug concentrations on CVF using regression modelling with a variable selection procedure. Following a total of 4291 person-years, the unadjusted CVF incidence rate was 0.54 per 100 person-years; 23 CVFs were reported (1.4% of 1651 individuals in these studies).
The BFA demonstrated rilpivirine resistance mutations (incidence rate ratio IRR=21.65, p<0.0001), HIV-1 subtype A6/A1 (IRR=12.87, p<0.0001), and body mass index (IRR=1.09 per 1 unit increase, p=0.04; IRR=3.97 of ≥30 kg/m2, p=0.01) were associated with CVF. Other variables including Q4W or Q8W dosing, female gender, or CAB/INSTI resistance mutations had no significant association with CVF. A combination of at least 2 of the following key baseline factors was associated with an increased risk of CVF: rilpivirine resistance mutations, HIV-1 subtype A6/A1, or BMI≥30 kg/m2 (see Table 6).
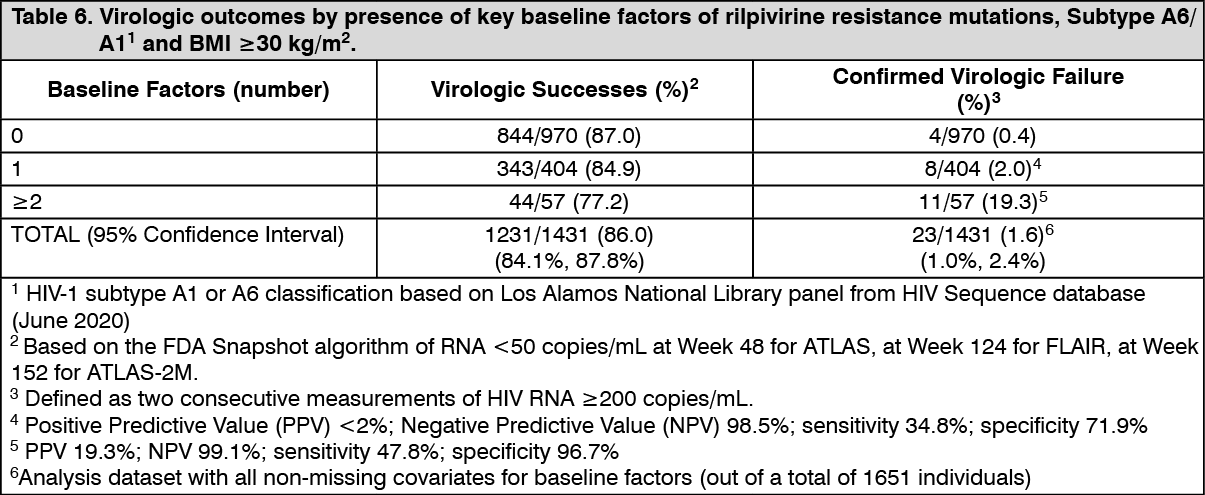 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn patients with at least two of these risk factors, the proportion of subjects who had a CVF was higher than observed in patients with none or one risk factor, with CVF identified in 6/24 patients [25.0%, 95%CI (9.8%, 46.7%)] treated with the every 2 months dosing regimen and 5/33 patients [15.2%, 95%CI (5.1%, 31.9%)] treated with the monthly dosing regimen.
Oral bridging with other ART: In a retrospective analysis of pooled data from 3 clinical studies (FLAIR, ATLAS-2M, and LATTE-2/study 200056), 29 subjects were included who received oral bridging for a median duration of 59 days (25th and 75th percentile 53-135) with ART other than Vocabria plus rilpivirine (alternative oral bridging) during treatment with Vocabria plus rilpivirine long-acting (LA) intramuscular (IM) injections. The median age of subjects was 32 years, 14% were female, 31% were non-white, 97% received an integrase inhibitor (INI)-based regimen for alternative oral bridging, 41% received an NNRTI as part of their alternative oral bridging regimen (including rilpivirine in 11/12 cases), and 62% received an NRTI. Three subjects withdrew during oral bridging or shortly following oral bridging for non-safety reasons. The majority (≥96%) of subjects maintained virologic suppression (plasma HIV-1 RNA <50 c/mL). During bridging with alternative oral bridging and during the period following alternative oral bridging (up to 2 Vocabria plus rilpivirine injections following oral bridging), no cases of CVF (plasma HIV-1 RNA ≥200 c/mL) were observed.
Paediatric population: The European Medicines Agency has deferred the obligation to submit the results of studies with Vocabria tablets/injection in one or more subsets of the paediatric population in the treatment of HIV-1 infection.
Pharmacokinetics: Cabotegravir pharmacokinetics is similar between healthy and HIV-infected subjects.
Tablet: The PK variability of cabotegravir is moderate. In Phase I studies in healthy subjects, between-subject CVb% for AUC, Cmax, and Ctau ranged from 26 to 34% across healthy subject studies and 28 to 56% across HIV-1 infected subject studies. Within-subject variability (CVw%) is lower than between-subject variability. (See Table 7.)
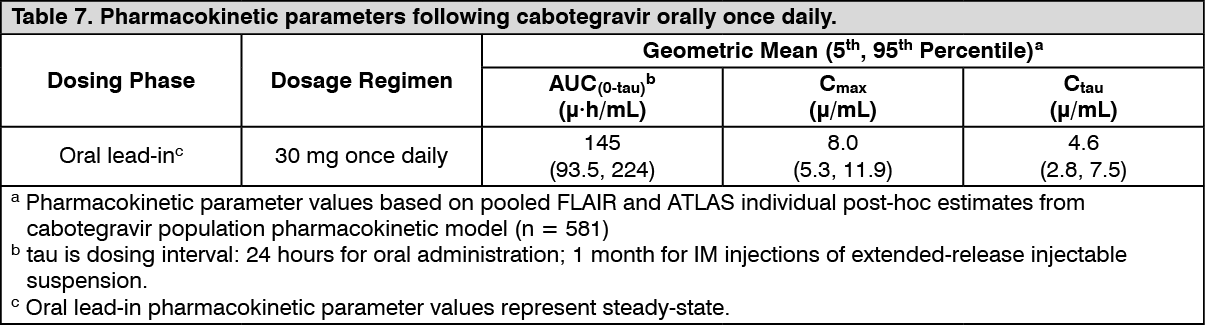 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSuspension for injection: The PK variability of cabotegravir is moderate to high. In HIV-infected subjects participating in Phase III studies, between-subject CVb% for Ctau ranged from 39 to 48%. Higher between-subject variability ranging from 41% to 89% was observed with single dose administration of long-acting cabotegravir injection. (See Table 8.)
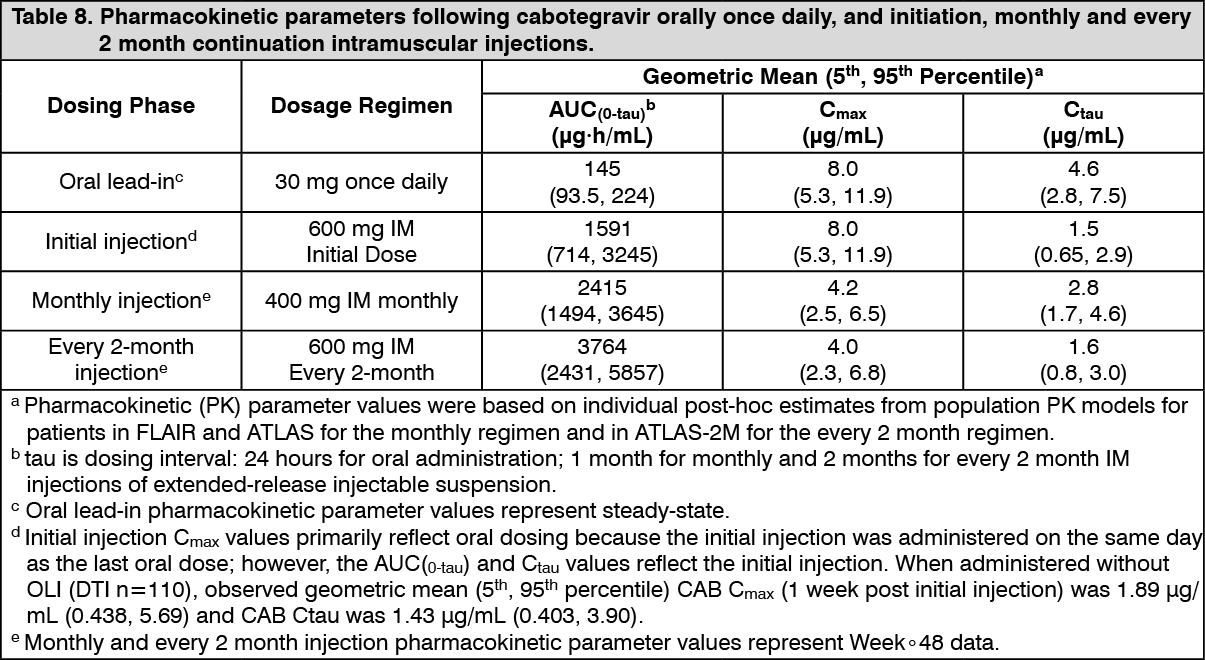 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAbsorption: Tablet: Cabotegravir is rapidly absorbed following oral administration, with median Tmax at 3 hours post dose for tablet formulation. With once daily dosing, pharmacokinetic steady-state is achieved by 7 days. Cabotegravir may be administered with or without food. Food increased the extent of absorption of cabotegravir. Bioavailability of cabotegravir is independent of meal content: high fat meals increased cabotegravir AUC(0-∞) by 14% and increased Cmax by 14% relative to fasted conditions. These increases are not clinically significant.
The absolute bioavailability of cabotegravir has not been established.
Suspension for injection: Cabotegravir injection exhibits absorption-limited (flip-flop) kinetics resulting from slow absorption from the gluteal muscle into the systemic circulation resulting in sustained plasma concentrations. Following a single intramuscular dose, plasma cabotegravir concentrations are detectable on the first day and gradually rise to reach maximum plasma concentration with a median Tmax of 7 days. Cabotegravir has been detected in plasma up to 52 weeks or longer after administration of a single injection. Pharmacokinetic steady-state is achieved by 44 weeks.
Plasma cabotegravir exposure increases in proportion or slightly less than in proportion to dose following single and repeat IM injection of doses ranging from 100 to 800 mg.
Distribution: Cabotegravir is highly bound (>99%) to human plasma proteins, based on in vitro data. Following administration of oral tablets, the mean apparent oral volume of distribution (Vz/F) in plasma was 12.3 L. In humans, the estimate of plasma cabotegravir Vc/F was 5.27 L and Vp/F was 2.43 L. These volume estimates, along with the assumption of high bioavailability, suggest some distribution of cabotegravir to the extracellular space.
Cabotegravir is present in the female and male genital tract. Median cervical and vaginal tissue: plasma ratios ranged from 0.16 to 0.28 and median rectal tissue: plasma ratios were ≤0.08 following a single 400 mg intramuscular injection (IM) at 4, 8, and 12 weeks after dosing.
Cabotegravir is present in cerebrospinal fluid (CSF). In HIV-infected subjects receiving a regimen of cabotegravir injection plus rilpivirine injection, the cabotegravir CSF to plasma concentration ratio [median (range)] (n=16) was 0.003 (range: 0.002 to 0.004) one week following a steady-state long acting cabotegravir (Q4W or Q8W) injection. Consistent with therapeutic cabotegravir concentrations in the CSF, CSF HIV-1 RNA (n=16) was <50 c/mL in 100% and <2 c/mL in 15/16 (94%) of subjects.
At the same time point, plasma HIV-1 RNA (n=18) was <50 c/mL in 100% and <2 c/mL in 12/18 (66.7%) of subjects.
In vitro, cabotegravir was not a substrate of organic anion transporting polypeptide (OATP) 1B1, OATP2B1 (Suspension for injection), OATP1B3 or organic cation transporter (OCT1).
Biotransformation: Cabotegravir is primarily metabolised by UGT1A1 with a minor UGT1A9 component. Cabotegravir is the predominant circulating compound in plasma, representing >90% of plasma total radiocarbon. Following oral administration in humans, cabotegravir is primarily eliminated through metabolism; renal elimination of unchanged cabotegravir is low (<1% of the dose). Forty-seven percent of the total oral dose is excreted as unchanged cabotegravir in the faeces. It is unknown if all or part of this is due to unabsorbed drug or biliary excretion of the glucuronide conjugate, which can be further degraded to form the parent compound in the gut lumen. Cabotegravir was observed to be present in duodenal bile samples. The glucuronide metabolite was also present in some, but not all, of the duodenal bile samples. Twenty-seven percent of the total oral dose is excreted in the urine, primarily as a glucuronide metabolite (75% of urine radioactivity, 20% of total dose).
Cabotegravir is not a clinically relevant inhibitor of the following enzymes and transporters: CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B4, UGT2B7, UGT2B15, and UGT2B17, P-gp, BCRP, Bile salt export pump (BSEP), OCT1, OCT2, OATP1B1, OATP1B3, multidrug and toxin extrusion transporter (MATE) 1, MATE 2-K, multidrug resistance protein (MRP) 2 or MRP4.
Elimination: Tablet: Cabotegravir has a mean terminal half-life of 41 h and an apparent clearance (CL/F) of 0.21 L per hour.
Suspension for injection: Cabotegravir mean apparent terminal phase half-life is absorption-rate limited and is estimated to be 5.6 to 11.5 weeks after a single dose IM injection. The significantly longer apparent half-life compared to oral reflects elimination from the injection site into the systemic circulation. The apparent CL/F was 0.151 L/h.
Linearity/non-linearity: Suspension for injection: Plasma CAB exposure increases in proportion or slightly less than in proportion to dose following single and repeat IM injection of doses ranging from 100 to 800 mg.
Polymorphisms: Tablet: In a meta-analysis of healthy and HIV-infected subject trials, subjects with UGT1A1 genotypes conferring poor cabotegravir metabolism had a 1.3- to 1.5-fold mean increase in steady-state cabotegravir AUC, Cmax, and Ctau compared with subjects with genotypes associated with normal metabolism via UGT1A1. These differences are not considered clinically relevant. No dose adjustment is required in subjects with UGT1A1 polymorphisms.
Suspension for injection: In a meta-analysis of healthy and HIV-infected subject trials, HIV-infected subjects with UGT1A1 genotypes conferring poor cabotegravir metabolism had a 1.2-fold mean increase in steady-state cabotegravir AUC, Cmax, and Ctau following long acting injection administration compared with subjects with genotypes associated with normal metabolism via UGT1A1. These differences are not considered clinically relevant. No dose adjustment is required in subjects with UGT1A1 polymorphisms.
Special patient populations: Gender: Population pharmacokinetic analyses revealed no clinically relevant effect of gender on the exposure of cabotegravir, therefore no dose adjustment is required on the basis of gender.
Race: Population pharmacokinetic analyses revealed no clinically relevant effect of race on the exposure of cabotegravir, therefore no dosage adjustment is required on the basis of race.
Body Mass Index (BMI): Population pharmacokinetic analyses revealed no clinically relevant effect of BMI on the exposure of cabotegravir, therefore no dose adjustment is required on the basis of BMI.
Elderly: Population pharmacokinetic analysis of cabotegravir revealed no clinically relevant effect of age on cabotegravir exposure. Pharmacokinetic data for cabotegravir in subjects of >65 years old are limited.
Renal impairment: No clinically important pharmacokinetic differences between subjects with severe renal impairment (CrCL <30 mL/min and not on dialysis) and matching healthy subjects were observed. No dosage adjustment is necessary for patients with mild to severe renal impairment (not on dialysis). Cabotegravir has not been studied in patients on dialysis.
Hepatic impairment: No clinically important pharmacokinetic differences between subjects with moderate hepatic impairment and matching healthy subjects were observed. No dosage adjustment is necessary for patients with mild to moderate hepatic impairment (Child-Pugh Score A or B). The effect of severe hepatic impairment (Child-Pugh Score C) on the pharmacokinetics of cabotegravir has not been studied.
Toxicology: Preclinical safety data: Carcinogenesis and mutagenesis: Cabotegravir was not mutagenic or clastogenic using in vitro tests in bacteria and cultured mammalian cells, and an in vivo rodent micronucleus assay. Cabotegravir was not carcinogenic in long term studies in the mouse and rat.
Reproductive toxicology studies: No effect on male or female fertility was observed in rats treated with cabotegravir at oral doses up to 1,000 mg/kg/day (>20 times the exposure in humans at the maximum recommended dose).
In an embryo-foetal development study there were no adverse developmental outcomes following oral administration of cabotegravir to pregnant rabbits up to a maternal toxic dose of 2,000 mg/kg/day (0.66 times the exposure in humans at the MRHD) or to pregnant rats at doses up to 1,000 mg/kg/day (>30 times the exposure in humans at the MRHD). In rats, alterations in foetal growth (decreased body weights) were observed at 1,000 mg/kg/day. Studies in pregnant rats showed that cabotegravir crosses the placenta and can be detected in foetal tissue.
In rat pre- and post-natal (PPN) studies cabotegravir reproducibly induced a delayed onset of parturition, and an increase in the number of stillbirths and neonatal mortalities at 1,000 mg/kg/day (>30 times the exposure in humans at the MRHD). A lower dose of 5 mg/kg/day (approximately 10 times the exposure in humans at the MRHD) cabotegravir was not associated with delayed parturition or neonatal mortality. In rabbit and rat studies there was no effect on survival when foetuses were delivered by caesarean section. Given the exposure ratio, the relevance to humans is unknown.
Repeated dose toxicity: The effect of prolonged daily treatment with high doses of cabotegravir has been evaluated in repeat oral dose toxicity studies in rats (26 weeks) and in monkeys (39 weeks). There were no drug-related adverse effects in rats or monkeys given cabotegravir orally at doses up to 1,000 mg/kg/day or 500 mg/kg/day, respectively.
In a 14 day and 28 day monkey toxicity study, gastro-intestinal (GI) effects (body weight loss, emesis, loose/watery faeces, and moderate to severe dehydration) were observed and were the result of local drug administration and not systemic toxicity.
In a 3 month study in rats, when cabotegravir was administered by monthly sub-cutaneous (SC) injection (up to 100 mg/kg/dose); monthly IM injection (up to 75 mg/kg/dose) or weekly SC injection (100 mg/kg/dose), there were no adverse effects noted and no new target organ toxicities (at exposures >30 times the exposure in humans at the MRHD of 400 mg IM dose).