Pharmacotherapeutic group: Endocrine therapy, Antiestrogens.
ATC code: L02BA03.
Pharmacology: Pharmacodynamics: Mechanism of action and pharmacodynamic effects: Fulvestrant is a competitive estrogen receptor (ER) antagonist with an affinity comparable to estradiol. Fulvestrant blocks the trophic actions of estrogens without any partial agonist (estrogen-like) activity. The mechanism of action is associated with downregulation of estrogen receptor protein levels. Clinical studies in postmenopausal women with primary breast cancer have shown that fulvestrant significantly downregulates ER protein in ER positive tumours compared with placebo. There was also a significant decrease in progesterone receptor expression consistent with a lack of intrinsic estrogen agonist effects. It has also been shown that fulvestrant 500 mg downregulates ER and the proliferation marker Ki67, to a greater degree than fulvestrant 250 mg in breast tumours in postmenopausal neoadjuvant setting.
Clinical efficacy and safety in advanced breast cancer: Two phase III clinical trials Studies 9238IL/0020 and 9238IL/0021 were completed in a total of 851 postmenopausal women with advanced breast cancer who had disease recurrence on or after adjuvant endocrine therapy or progression following endocrine therapy for advanced disease. 77% of the study population had estrogen receptor positive breast cancer. These trials compared the safety and efficacy of monthly administration of Faslodex 250 mg versus the daily administration of 1 mg anastrozole (aromatase inhibitor). Overall, Faslodex at the 250 mg monthly dose was at least as effective as anastrozole in terms of progression-free survival, objective response, and time to death. There were no statistically significant differences in any of these endpoints between the two treatment groups. Progression-free survival was the primary endpoint. Combined analysis of both trials showed that 83% of patients who received Faslodex progressed, compared with 85% of patients who received anastrozole. Combined analysis of both trials showed the hazard ratio of Faslodex 250 mg to anastrozole for progression-free survival was 0.95 (95% CI 0.82 to 1.10). The objective response rate for Faslodex 250 mg was 19.2% compared with 16.5% for anastrozole. The median time to death was 27.4 months for patients treated with Faslodex and 27.6 months for patients treated with anastrozole. The hazard ratio of Faslodex 250 mg to anastrozole for time to death was 1.01 (95% CI 0.86 to 1.19).
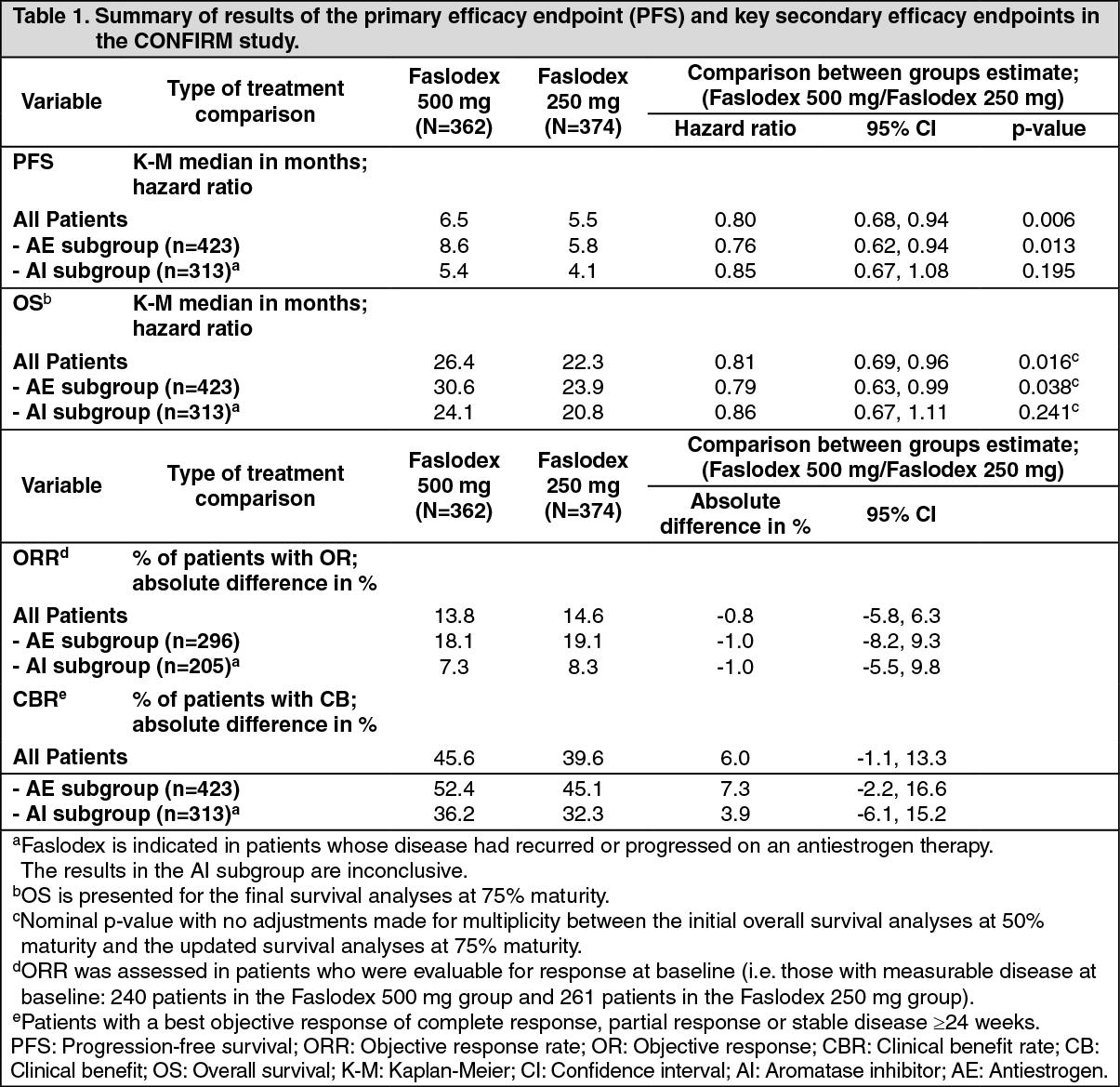
A Phase 3 clinical study was completed in 736 postmenopausal women with advanced breast cancer who had disease recurrence on or after adjuvant endocrine therapy or progression following endocrine therapy for advanced disease. The study included 423 patients whose disease had recurred or progressed during antiestrogen therapy (AE subgroup) and 313 patients whose disease had recurred or progressed during aromatase inhibitor therapy (AI subgroup). This study compared the efficacy and safety of Faslodex 500 mg (n=362) with Faslodex 250 mg (n=374). Progression-free survival (PFS) was the primary endpoint; key secondary efficacy endpoints included objective response rate (ORR), clinical benefit rate (CBR) and overall survival (OS). Efficacy results for the CONFIRM study are summarized in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
A Phase 3, randomised, double-blind, double-dummy, multicentre study of Faslodex 500 mg versus anastrozole 1 mg was conducted in postmenopausal women with ER-positive and/or PgR-positive locally advanced or metastatic breast cancer who had not previously been treated with any hormonal therapy. A total of 462 patients were randomised 1:1 sequentially to receive either fulvestrant 500 mg or anastrozole 1 mg.
Randomisation was stratified by disease setting (locally advanced or metastatic), prior chemotherapy for advanced disease, and measurable disease.
The primary efficacy endpoint of the study was investigator assessed progression-free survival (PFS) evaluated according to RECIST 1.1 (Response Evaluation Criteria in Solid Tumours). Key secondary efficacy endpoints included overall survival (OS) and objective response rate (ORR).
Patients enrolled in this study had a median age of 63 years (range 36-90). The majority of patients (87.0%) had metastatic disease at baseline. Fifty-five percent (55.0%) of patients had visceral metastasis at baseline. A total of 17.1% of patients received a prior chemotherapy regimen for advanced disease; 8% of patients had measurable disease.
Consistent results were observed across the majority of pre-specified patient subgroups. For the subgroup of patients with disease limited to non-visceral metastasis (n=208), the HR was 0.592 (95% CI: 0.419, 0.837) for the Faslodex arm compared to the anastrozole arm. For the subgroup of patients with visceral metastasis (n=254), the HR was 0.993 (95% CI: 0.740, 1.331) for the Faslodex arm compared to the anastrozole arm. The efficacy results of the FALCON study are presented in Table 2 and figure. (See Table 2 and figure).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Two Phase 3 clinical studies were completed in a total of 851 postmenopausal women with advanced breast cancer who had disease recurrence on or after adjuvant endocrine therapy or progression following endocrine therapy for advanced disease. Seventy seven percent (77%) of the study population had estrogen receptor positive breast cancer. These studies compared the safety and efficacy of monthly administration of Faslodex 250 mg versus the daily administration of 1 mg anastrozole (aromatase inhibitor). Overall, Faslodex at the 250 mg monthly dose was at least as effective as anastrozole in terms of progression-free survival, objective response, and time to death. There were no statistically significant differences in any of these endpoints between the two treatment groups. Progression-free survival was the primary endpoint. Combined analysis of both studies showed that 83% of patients who received Faslodex progressed, compared with 85% of patients who received anastrozole. Combined analysis of both studies showed the hazard ratio of Faslodex 250 mg to anastrozole for progression-free survival was 0.95 (95% CI 0.82 to 1.10). The objective response rate for Faslodex 250 mg was 19.2% compared with 16.5% for anastrozole. The median time to death was 27.4 months for patients treated with Faslodex and 27.6 months for patients treated with anastrozole. The hazard ratio of Faslodex 250 mg to anastrozole for time to death was 1.01 (95% CI 0.86 to 1.19).
Effects on the postmenopausal endometrium: Preclinical data do not suggest a stimulatory effect of fulvestrant on the postmenopausal endometrium (see Toxicology: Preclinical safety data as follows). A 2-week study in healthy postmenopausal volunteers treated with 20 μg per day ethinylestradiol showed that pretreatment with Faslodex 250 mg resulted in significantly reduced stimulation of the postmenopausal endometrium, compared to pre-treatment with placebo, as judged by ultrasound measurement of endometrium thickness.
Neoadjuvant treatment for up to 16 weeks in breast cancer patients treated with either Faslodex 500 mg or Faslodex 250 mg did not result in clinically significant changes in endometrial thickness, indicating a lack of agonist effect. There is no evidence of adverse endometrial effects in the breast cancer patients studied. No data are available regarding endometrial morphology.
In two short-term studies (1 and 12 weeks) in premenopausal patients with benign gynaecologic disease, no significant differences in endometrial thickness were observed by ultrasound measurement between fulvestrant and placebo groups.
Effects on bone: There are no long-term data on the effect of fulvestrant on bone. Neoadjuvant treatment for up to 16 weeks in breast cancer patients with either Faslodex 500 mg or Faslodex 250 mg did not result in clinically significant changes in serum bone-turnover markers.
Paediatric population: Faslodex is not indicated for use in children. The European Medicines Agency has waived the obligation to submit the results of studies with Faslodex in all subsets of the paediatric population in breast cancer (see Dosage & Administration for information on paediatric use).
An open-label Phase 2 study investigated the safety, efficacy and pharmacokinetics of fulvestrant in 30 girls aged 1 to 8 years with Progressive Precocious Puberty associated with McCune Albright Syndrome (MAS). The paediatric patients received 4 mg/kg monthly intramuscular dose of fulvestrant. This 12-month study investigated a range of MAS endpoints and showed a reduction in the frequency of vaginal bleeding and a reduction in the rate of bone age advancement. The steady-state trough concentrations of fulvestrant in children in this study were consistent in adults (see Pharmacokinetics as follows). There were no new safety concerns arising from this small study, but 5-year data are yet not available.
Pharmacokinetics: Absorption: After administration of Faslodex long-acting intramuscular injection, fulvestrant is slowly absorbed and maximum plasma concentrations (C
max) are reached after about 5 days. Administration of Faslodex 500 mg regimen achieves exposure levels at, or close to, steady state within the first month of dosing (mean [CV]: AUC 475 [33.4%] ng.days/ml, C
max 2 [35.3%] ng/ml, C
min 16.3 [25.9%] ng/ml, respectively). At steady state, fulvestrant plasma concentrations are maintained within a relatively narrow range with up to an approximately 3-fold difference between maximum and trough concentrations. After intramuscular administration, the exposure is approximately dose-proportional in the dose range 50 to 500 mg.
Distribution: Fulvestrant is subject to extensive and rapid distribution. The large apparent volume of distribution at steady state (Vd
ss) of approximately 3 to 5 l/kg suggests that distribution is largely extravascular. Fulvestrant is highly (99%) bound to plasma proteins. Very low density lipoprotein (VLDL), low density lipoprotein (LDL), and high density lipoprotein (HDL) fractions are the major binding components. No interaction studies were conducted on competitive protein binding. The role of sex hormone-binding globulin (SHBG) has not been determined.
Biotransformation: The metabolism of fulvestrant has not been fully evaluated, but involves combinations of a number of possible biotransformation pathways analogous to those of endogenous steroids. Identified metabolites (includes 17-ketone, sulphone, 3-sulphate, 3- and 17-glucuronide metabolites) are either less active or exhibit similar activity to fulvestrant in antiestrogen models. Studies using human liver preparations and recombinant human enzymes indicate that CYP3A4 is the only P450 isoenzyme involved in the oxidation of fulvestrant; however, non-P450 routes appear to be more predominant
in vivo.
In vitro data suggest that fulvestrant does not inhibit CYP450 isoenzymes.
Elimination: Fulvestrant is eliminated mainly in metabolised form. The major route of excretion is via the faeces, with less than 1% being excreted in the urine. Fulvestrant has a high clearance, 11±1.7 ml/min/kg, suggesting a high hepatic extraction ratio. The terminal half-life (t
½) after intramuscular administration is governed by the absorption rate and was estimated to be 50 days.
Special populations: In a population pharmacokinetic analysis of data from Phase 3 studies, no difference in fulvestrant's pharmacokinetic profile was detected with regard to age (range 33 to 89 years), weight (40-127 kg) or race.
Renal impairment: Mild to moderate impairment of renal function did not influence the pharmacokinetics of fulvestrant to any clinically relevant extent.
Hepatic impairment: The pharmacokinetics of fulvestrant has been evaluated in a single-dose clinical study conducted in women with mild to moderate hepatic impairment (Child-Pugh class A and B). A high dose of a shorter duration intramuscular injection formulation was used. There was up to about 2.5-fold increase in AUC in women with hepatic impairment compared to healthy subjects. In patients administered Faslodex, an increase in exposure of this magnitude is expected to be well tolerated. Women with severe hepatic impairment (Child-Pugh class C) were not evaluated.
Paediatric population: The pharmacokinetics of fulvestrant has been evaluated in a clinical study conducted in 30 girls with Progressive Precocious Puberty associated with McCune Albright Syndrome. The paediatric patients were aged 1 to 8 years and received 4 mg/kg monthly intramuscular dose of fulvestrant. The geometric mean (standard deviation) steady state trough concentration (C
min,ss) and AUC
ss was (0.9) ng/mL and 3680 (1020) ng*hr/mL, respectively. Although the data collected were limited, the steady-state trough concentrations of fulvestrant in children appear to be consistent with those in adults.
Toxicology: Preclinical safety data: The acute toxicity of fulvestrant is low.
Faslodex and other formulations of fulvestrant were well tolerated in animal species used in multiple dose studies. Local reactions, including myositis and granulomata at the injection site were attributed to the vehicle but the severity of myositis in rabbits increased with fulvestrant, compared to the saline control. In toxicity studies with multiple intramuscular doses of fulvestrant in rats and dogs, the antiestrogenic activity of fulvestrant was responsible for most of the effects seen, particularly in the female reproductive system, but also in other organs sensitive to hormones in both sexes. Arteritis involving a range of different tissues was seen in some dogs after chronic (12 months) dosing.
In dog studies following oral and intravenous administration, effects on the cardiovascular system (slight elevations of the S-T segment of the ECG [oral], and sinus arrest in one dog [intravenous]) were seen. These occurred at exposure levels higher than in patients (C
max >15 times) and are likely to be of limited significance for human safety at the clinical dose.
Fulvestrant showed no genotoxic potential.
Fulvestrant showed effects upon reproduction and embryo/foetal development consistent with its antiestrogenic activity, at doses similar to the clinical dose. In rats, a reversible reduction in female fertility and embryonic survival, dystocia and an increased incidence of foetal abnormalities including tarsal flexure were observed. Rabbits given fulvestrant failed to maintain pregnancy. Increases in placental weight and post-implantation loss of foetuses were seen. There was an increased incidence 10 of foetal variations in rabbits (backwards displacement of the pelvic girdle and 27 pre-sacral vertebrae).
A two-year oncogenicity study in rats (intramuscular administration of Faslodex) showed increased incidence of ovarian benign granulosa cell tumours in female rats at the high dose, 10 mg/rat/15 days and an increased incidence of testicular Leydig cell tumours in males. In a two-year mouse oncogenicity study (daily oral administration) there was an increased incidence of ovarian sex cord stromal tumours (both benign and malignant) at doses of 150 and 500 mg/kg/day. At the no-effect level for these findings, systemic exposure levels (AUC) were, in rats, approximately 1.5-fold the expected human exposure levels in females and 0.8-fold in males, and in mice, approximately 0.8-fold the expected human exposure levels in both males and females. Induction of such tumours is consistent with pharmacology-related endocrine feedback alterations in gonadotropin levels caused by antiestrogens in cycling animals. Therefore these findings are not considered to be relevant to the use of fulvestrant in postmenopausal women with
advanced breast cancer.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image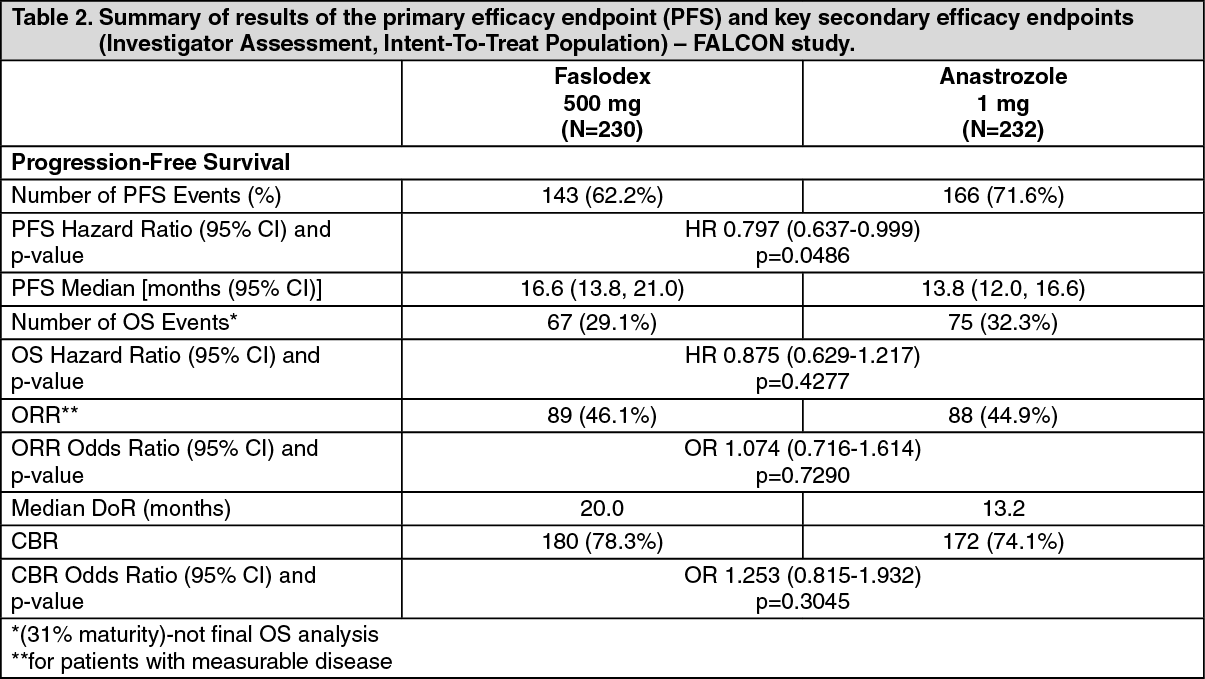 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image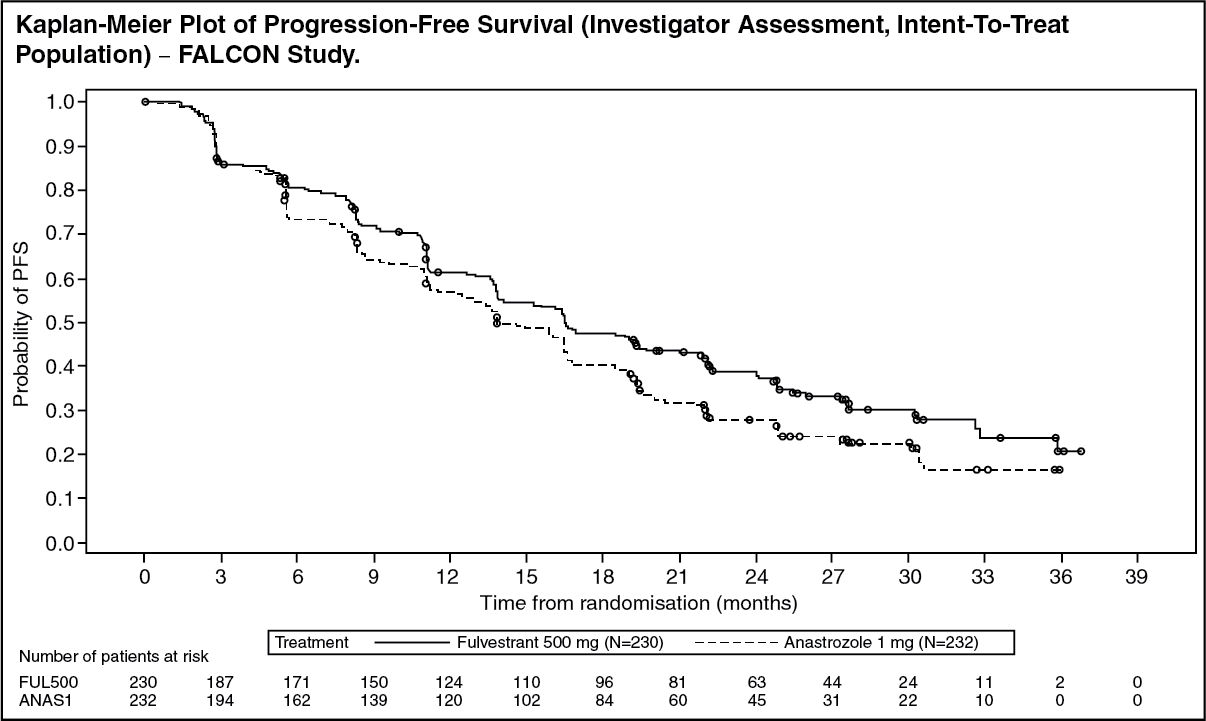 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out