


 Sign Out
Sign Out
The Grade ≥ 3 adverse reactions occurring in > 2% of patients were anaemia (16%), neutropenia (5%), fatigue/asthenia (5%), thrombocytopenia (3%) and leukopenia (2%).
Adverse reactions that most commonly led to dose interruptions and/ or reductions in monotherapy were anaemia (17%), fatigue/asthenia (6%), vomiting (6%), nausea (6%), and neutropenia (6%). Adverse reactions that most commonly led to permanent discontinuation were anaemia (1.8%), thrombocytopenia (0.8%), fatigue/asthenia (0.7%), nausea (0.6%), neutropenia (0.5%) and vomiting (0.5%).
When Lynparza is used in combination with bevacizumab the safety profile is generally consistent with that of the individual therapies.
Adverse events led to dose interruption and/ or reduction of olaparib in 57.4% of patients when used in combination with bevacizumab and led to permanent discontinuation of treatment with olaparib/bevacizumab and placebo/bevacizumab in 20.4% and 5.6% of patients, respectively. The adverse reactions that most commonly led to dose interruption and/or reduction were anaemia (21.5%), nausea (9.5%) and fatigue/asthenia (5.2%). The adverse reactions that most commonly led to permanent discontinuation were anaemia (3.6%), nausea (3.4%) and fatigue/asthenia (1.5%).
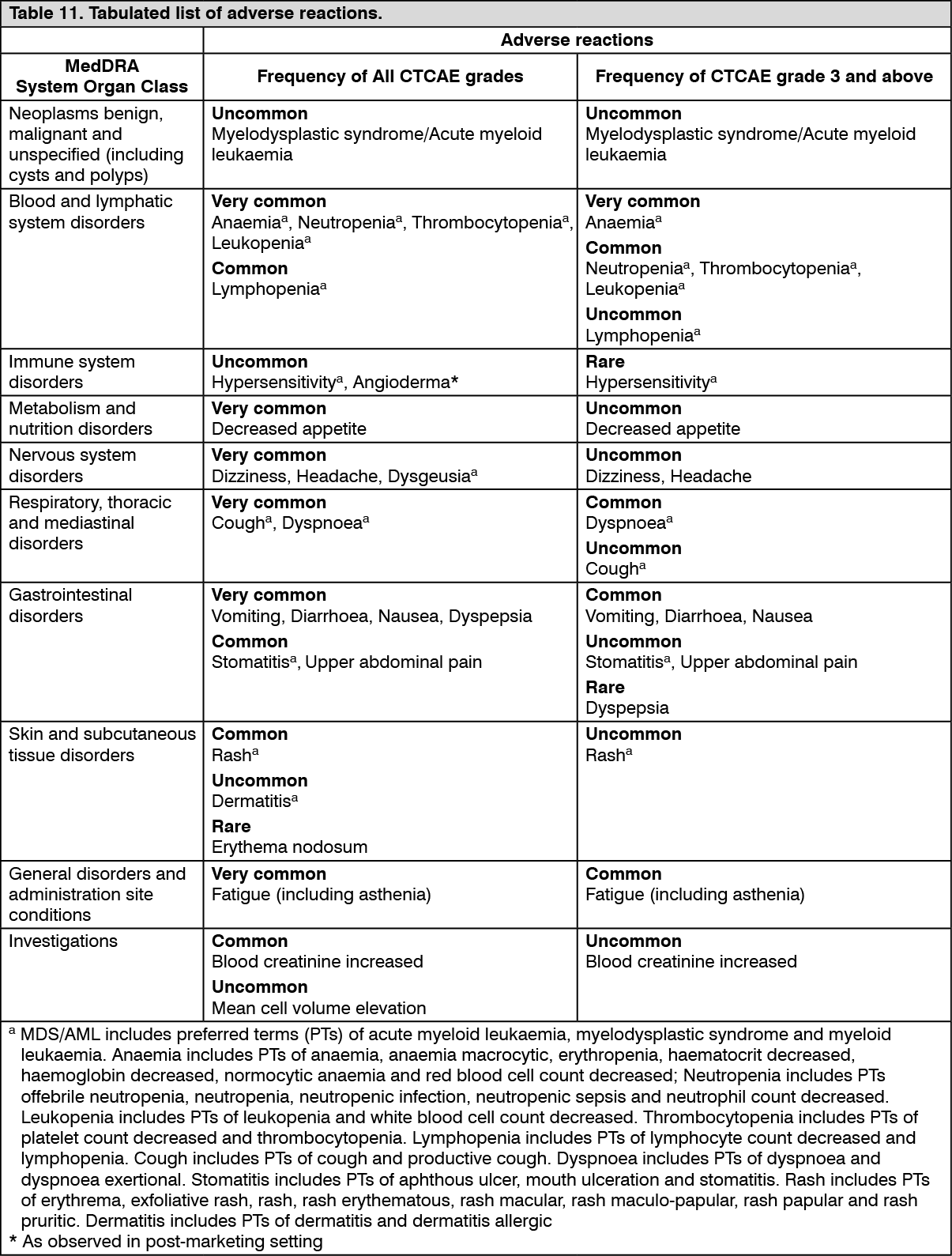
Tabulated list of adverse reactions: The safety profile is based on pooled data from 3077 patients with solid tumours treated with Lynparza monotherapy in clinical trials at the recommended dose.
The following adverse reactions have been identified in clinical trials with patients receiving Lynparza monotherapy where patient exposure is known. Adverse drug reactions are listed by MedDRA System Organ Class (SOC) and then by MedDRA preferred term in Table 11. Within each SOC, preferred terms are arranged by decreasing frequency and then by decreasing seriousness. Frequencies of occurrence of adverse reactions are defined as: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1000); very rare (<1/10,000); not known (cannot be estimated from available data). (See Table 11.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Myelodysplastic syndrome/Acute myeloid leukaemia: In clinical studies, across all indications, MDS/AML occurred uncommonly in patients on treatment and during the 30-day safety follow up, and <1.5% at any time after starting olaparib, including cases actively solicited during the long term follow up for overall survival. In patients with BRCAm platinum-sensitive relapsed ovarian cancer who had received at least two prior lines of platinum chemotherapy and received study treatment until disease progression (SOLO2 study, with olaparib treatment ≥ 2 years in 45% of patients), the incidence of MDS/AML was 8% in patients receiving olaparib and 4% in patients receiving placebo at a follow-up of 5 years. In the olaparib arm, 9 out of 16 MDS/AML cases occurred after discontinuation of olaparib during the survival follow-up. The incidence of MDS/AML was observed in the context of extended overall survival in the olaparib arm and late onset of MDS/AML. The risk of MDS/AML remains < 1.5% at 5 year follow up in the first-line setting when olaparib maintenance treatment is given after one line of platinum chemotherapy for a duration of 2 years.
Haematological toxicity: Anaemia and other haematological toxicities were generally low grade (CTCAE grade 1 or 2), however, there were reports of CTCAE grade 3 and higher events. Anaemia was the most common CTCAE grade ≥3 adverse reaction reported in clinical studies. Median time to first onset of anaemia was approximately 4 weeks (approximately 7 weeks for CTCAE grade ≥3 events). Anaemia was managed with dose interruptions and dose reductions (see Dosage & Administration), and where appropriate with blood transfusions. In clinical studies with the tablet formulation, the incidence of anaemia adverse reactions was 39.2% (CTCAE grade ≥3 17.2%) and the incidences of dose interruptions, reductions and discontinuations for anaemia were 17.8%, 11.1% and 2.2%, respectively; 21.8% of patients treated with olaparib needed one or more blood transfusions. An exposure-response relationship between olaparib and decreases in haemoglobin has been demonstrated. In clinical studies with Lynparza the incidence of CTCAE grade ≥ 2 shifts (decreases) from baseline in haemoglobin was 20%, absolute neutrophils 20%, platelets 5%, lymphocytes 30% and leucocytes 20% (all % approximate).
The incidence of elevations in mean corpuscular volume from low or normal at baseline to above the ULN was approximately 68%. Levels appeared to return to normal after treatment discontinuation and did not appear to have any clinical consequences.
Baseline testing, followed by monthly monitoring of complete blood counts is recommended for the first 12 months of treatment and periodically after this time to monitor for clinically significant changes in any parameter during treatment which may require dose interruption or reduction and/or further treatment (see Dosage & Administration and Precautions).
Other laboratory findings: In clinical studies with Lynparza the incidence of CTCAE grade ≥ 2 shifts (elevations) from baseline in blood creatinine was approximately 11%. Data from a double-blind placebo-controlled study showed median increase up to 23% from baseline remaining consistent over time and returning to baseline after treatment discontinuation, with no apparent clinical sequelae. 90% of patients had creatinine values of CTCAE grade 0 at baseline and 10% were CTCAE grade 1 at baseline.
Gastrointestinal toxicities: Nausea was generally reported very early, with first onset within the first month of Lynparza treatment in the majority of patients. Vomiting was reported early, with first onset within the first two months of Lynparza treatment in the majority of patients. Both nausea and vomiting were reported to be intermittent for the majority of patients and can be managed by dose interruption, dose reduction and/or antiemetic therapy. Antiemetic prophylaxis is not required.
In ovarian cancer maintenance treatment, patients experienced nausea events (77% on olaparib, 38% on placebo), vomiting (40% on olaparib, 15% on placebo), diarrhoea (34% on olaparib, 25% on placebo) and dyspepsia (17% on olaparib, 12% on placebo). Nausea events led to discontinuation in 2.3% of olaparib-treated patients (CTCAE Grade 2) and 0.8% of placebo-treated patients (CTCAE Grade 1); 0.8% and 0.4% of olaparib-treated patients discontinued treatment due to low grade (CTCAE Grade 2) vomiting and dyspepsia, respectively. No olaparib or placebo-treated patients discontinued due to diarrhoea. No placebo-treated patients discontinued due to vomiting or dyspepsia. Nausea events led to dose interruption and dose reductions in 14% and 4%, respectively, of olaparib-treated patients. Vomiting events led to interruption in 10% of olaparib-treated patients; no olaparib-treated patients experienced a vomiting event leading to dose reduction.
Paediatric population: No studies have been conducted in paediatric patients.
Other special populations: Limited safety data are available in non-Caucasian patients.
View ADR Monitoring Form