


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Mechanism of action: Pomalidomide has direct anti-myeloma tumoricidal activity, immunomodulatory activities and inhibits stromal cell support for multiple myeloma tumour cell growth. Specifically, pomalidomide inhibits proliferation and induces apoptosis of haematopoietic tumour cells. Additionally, pomalidomide inhibits the proliferation of lenalidomide-resistant multiple myeloma cell lines and synergises with dexamethasone in both lenalidomide-sensitive and lenalidomide-resistant cell lines to induce tumour cell apoptosis. Pomalidomide enhances T cell- and natural killer (NK) cell-mediated immunity and inhibits production of pro-inflammatory cytokines (e.g., TNF-α and IL-6) by monocytes. Pomalidomide also inhibits angiogenesis by blocking the migration and adhesion of endothelial cells.
Pomalidomide binds directly to the protein cereblon (CRBN), which is part of an E3 ligase complex that includes deoxyribonucleic acid (DNA) damage-binding protein 1(DDB1), cullin 4 (CUL4), and regulator of cullins-1 (Roc1), and can inhibit the auto-ubiquitination of CRBN within the complex. E3 ubiquitin ligases are responsible for the poly-ubiquitination of a variety of substrate proteins, and may partially explain the pleiotropic cellular effects observed with pomalidomide treatment.
In the presence of pomalidomide in vitro, substrate proteins Aiolos and Ikaros are targeted for ubiquitination and subsequent degradation leading to direct cytotoxic and immunomodulatory effects. In vivo, pomalidomide therapy led to reduction in the levels of Ikaros in patients with relapsed lenalidomide-refractory multiple myeloma.
Clinical efficacy and safety: Pomalidomide in combination with bortezomib and dexamethasone: The efficacy and safety of pomalidomide in combination with bortezomib and low-dose dexamethasone (Pom+Btz+LD-Dex) was compared with bortezomib and low-dose dexamethasone (Btz+LD-Dex) in a Phase III multi-centre, randomised, open-label study (CC-4047-MM-007), in previously treated adult patients with multiple myeloma, who had received at least one prior regimen, including lenalidomide and have demonstrated disease progression on or after the last therapy. A total of 559 patients were enrolled and randomised in the study: 281 in the Pom+Btz+LD-Dex arm and 278 in the Btz+LD-Dex arm. 54% of patients were male with median age for the overall population of 68 years (min, max: 27, 89 years).
Approximately 70% of patients were refractory to lenalidomide (71.2% in Pom+Btz+LD-Dex, 68.7% in Btz+LD-Dex). Approximately 40% of patients were in 1st relapse and approximately 73% of patients received bortezomib as prior treatment. Patients in the Pom+Btz+LD-Dex arm were administered 4 mg pomalidomide orally on Days 1 to 14 of each 21-day cycle. Bortezomib (1.3 mg/m2/dose) was administered to patients in both study arms on Days 1, 4, 8 and 11 of a 21-day cycle for Cycles 1 to 8; and on Days 1 and 8 of a 21-day cycle for Cycles 9 and onwards. Low-dose dexamethasone (20 mg/day [≤75 years old] or 10 mg/day [>75 years old]) was administered to patients in both study arms on Days 1, 2, 4, 5, 8, 9, 11 and 12 of a 21-day cycle for Cycles 1 to 8; and on Days 1, 2, 8 and 9 of each subsequent 21-day cycle from Cycles 9 onwards. Doses were reduced and treatment was temporarily interrupted or stopped as needed to manage toxicity (see Dosage & Administration).
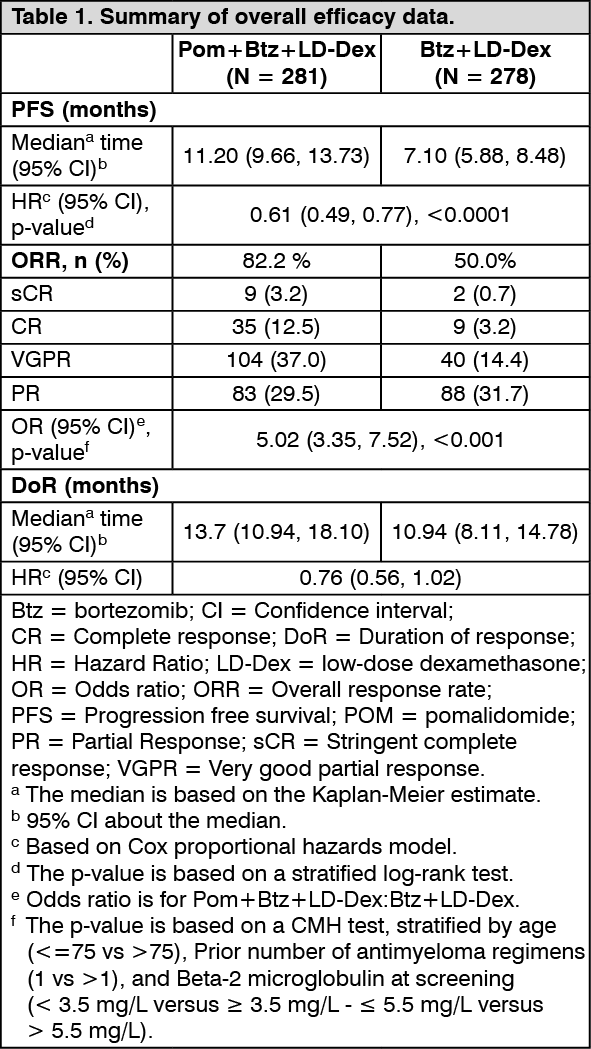
The primary efficacy endpoint was Progression Free Survival (PFS) assessed by an Independent Response Adjudication Committee (IRAC) according to the IMWG criteria using the intent to treat population (ITT). After a median follow-up of 15.9 months, median PFS time was 11.20 months (95% CI: 9.66, 13.73) in the Pom+Btz+LD-Dex arm. In the Btz+LD-Dex arm, median PFS time was 7.1 months (95% CI: 5.88, 8.48).
Summary of overall efficacy data are presented in Table 1 using a cut-off date of 26 Oct 2017. Kaplan-Meier curve for PFS for the ITT population is provided in Figure 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe median duration of treatment was 8.8 months (12 treatment cycles) in the Pom+Btz+LD-Dex arm and 4.9 months (7 treatment cycles) in the Btz+LD-Dex arm.
The PFS advantage was more pronounced in patients who received only one prior line of therapy. In patients who received 1 prior antimyeloma line, median PFS time was 20.73 months (95% CI: 15.11, 27.99) in the Pom+Btz+LD-Dex arm and 11.63 months (95% CI: 7.52, 15.74) in the Btz+LD-Dex arm. A 46% risk reduction was observed with Pom+Btz+LD-Dex treatment (HR = 0.54, 95% CI: 0.36, 0.82). (See Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAs per an interim analysis for Overall Survival (OS), using a cut-off of 15 September 2018 (median follow-up period of 26.2 months), median OS time from Kaplan-Meier estimates was 40.5 months for the Pom+Btz+LD-Dex arm and 30.5 months for the Btz+LD-Dex arm; HR = 0.91, 95% CI: 0.70, 1.18, with an overall event rate of 43.3%.
Pomalidomide in combination with dexamethasone: The efficacy and safety of pomalidomide in combination with dexamethasone were evaluated in a Phase III multi-centre, randomised, open-label study (CC-4047-MM-003), where pomalidomide plus low-dose dexamethasone therapy (Pom+LD-Dex) was compared to high-dose dexamethasone alone (HD-Dex) in previously treated adult patients with relapsed and refractory multiple myeloma, who have received at least two prior treatment regimens, including both lenalidomide and bortezomib, and have demonstrated disease progression on the last therapy. A total of 455 patients were enrolled in the study: 302 in the Pom+LD-Dex arm and 153 in the HD-Dex arm. The majority of patients were male (59%) and white (79%); the median age for the overall population was 64 years (min, max: 35, 87 years).
Patients in the Pom+LD-Dex arm were administered 4 mg pomalidomide orally on days 1 to 21 of each 28-day cycle. LD-Dex (40 mg) was administered once per day on days 1, 8, 15 and 22 of a 28-day cycle. For the HD-Dex arm, dexamethasone (40 mg) was administered once per day on days 1 through 4, 9 through 12, and 17 through 20 of a 28-day cycle. Patients >75 years of age started treatment with 20 mg dexamethasone. Treatment continued until patients had disease progression.
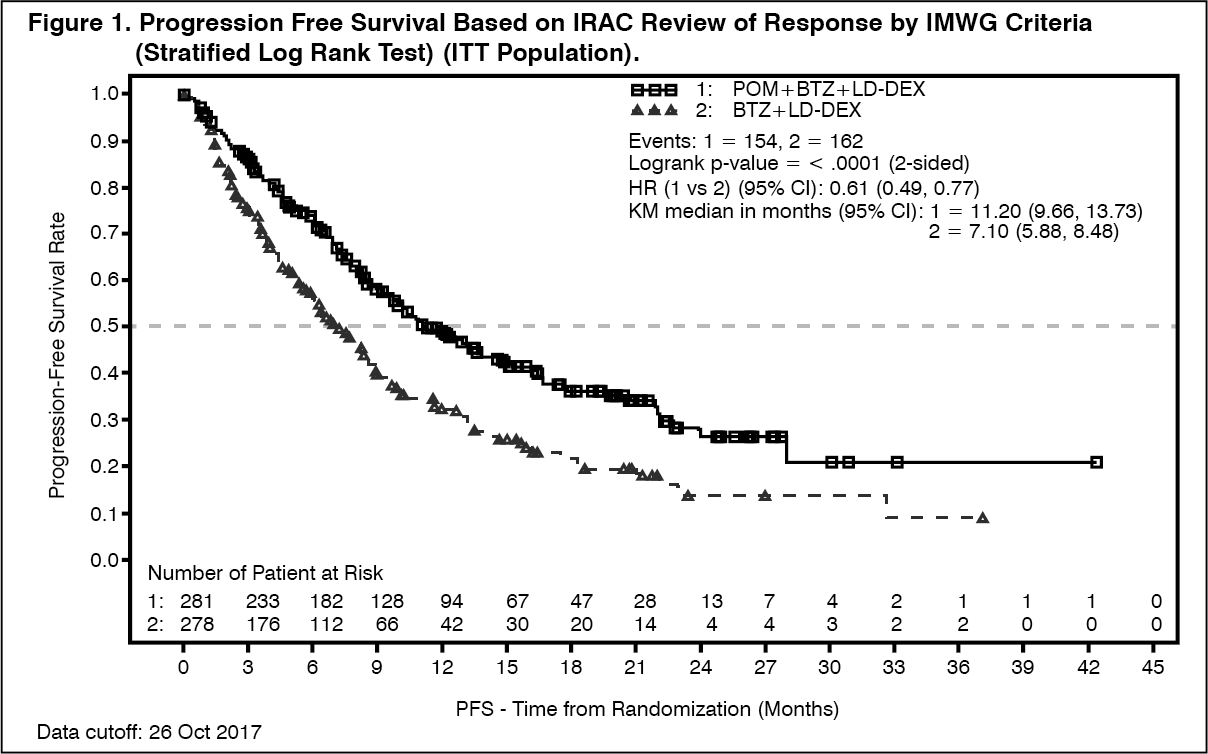
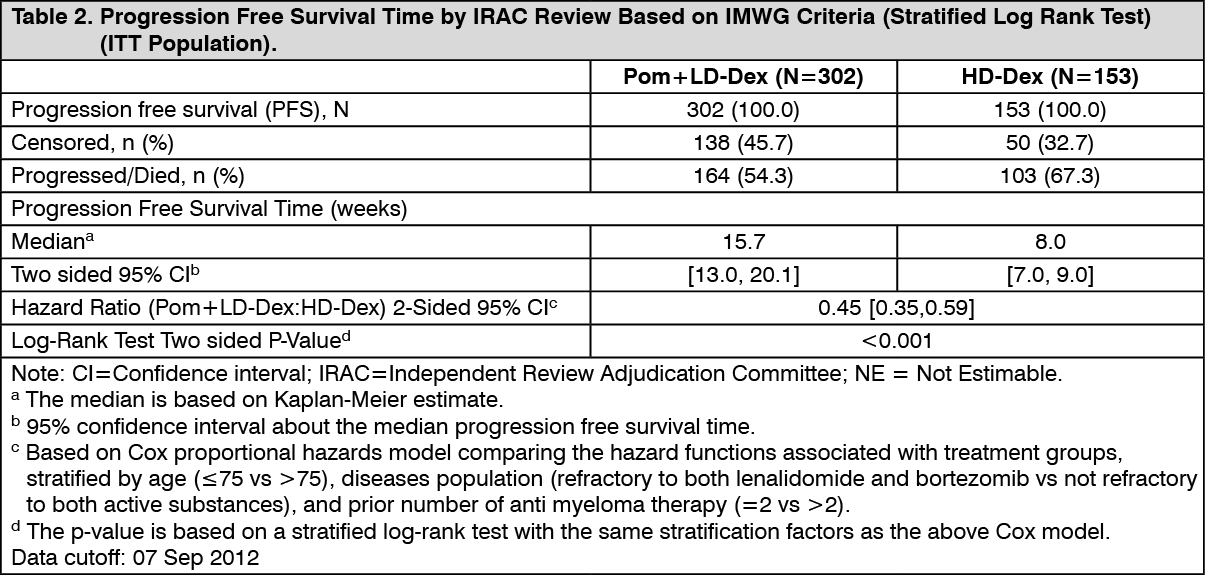
The primary efficacy endpoint was progression free survival by International Myeloma Working Group (IMWG criteria). For the intention to treat (ITT) population, median PFS time by Independent Review Adjudication Committee (IRAC) review based on IMWG criteria was 15.7 weeks (95% CI: 13.0, 20.1) in the Pom+LD-Dex arm; the estimated 26-week event-free survival rate was 35.99% (±3.46%). In the HD-Dex arm, median PFS time was 8.0 weeks (95% CI: 7.0, 9.0); the estimated 26-week event-free survival rate was 12.15% (±3.63%).
PFS was evaluated in several relevant subgroups: gender, race, ECOG performance status, stratification factors (age, disease population, prior anti-myeloma therapies [2, >2]), selected parameters of prognostic significance (baseline beta-2 microglobulin level, baseline albumin levels, baseline renal impairment, and cytogenetic risk), and exposure and refractoriness to prior anti-myeloma therapies. Regardless of the subgroup evaluated, PFS was generally consistent with that observed in the ITT population for both treatment groups.
PFS is summarised in Table 2 for the ITT population. Kaplan-Meier curve for PFS for the ITT population is provided in Figure 2. (See Table 2 and Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image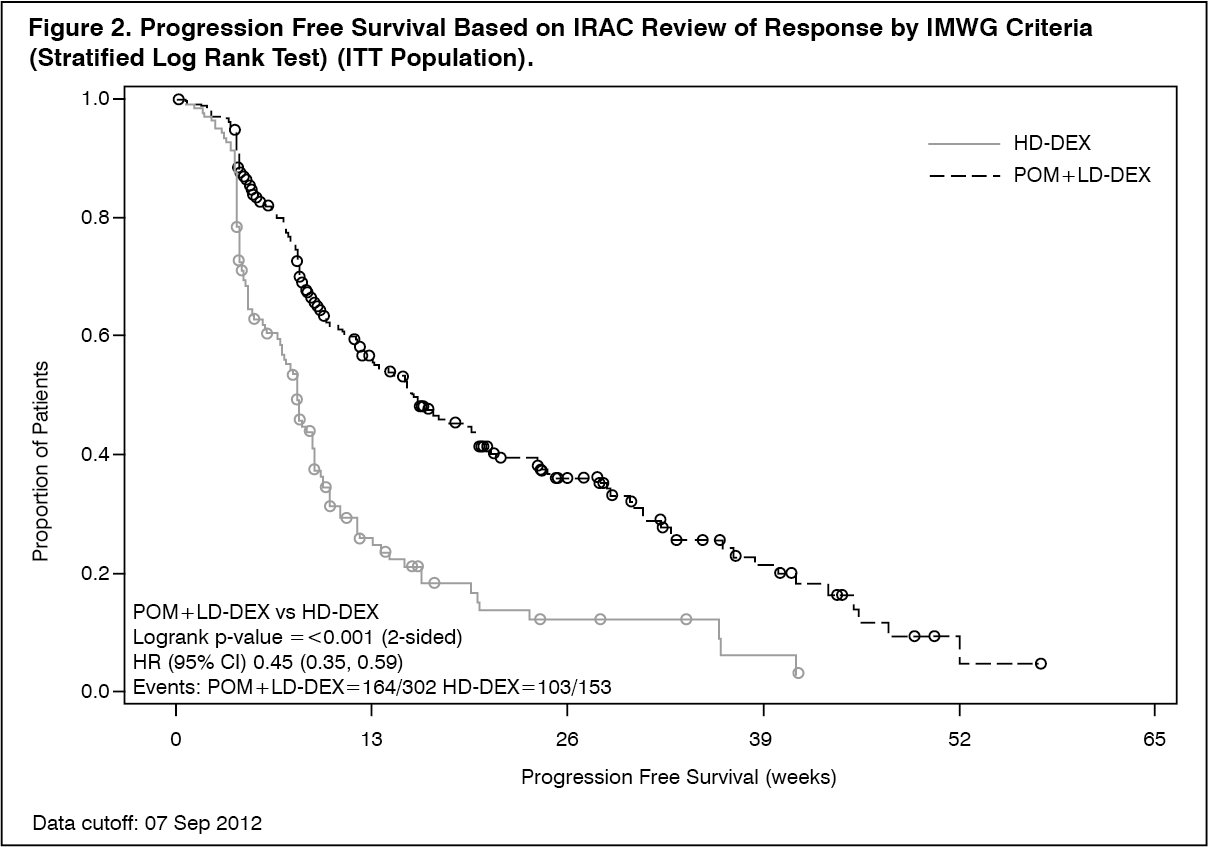 Click on icon to see table/diagram/image
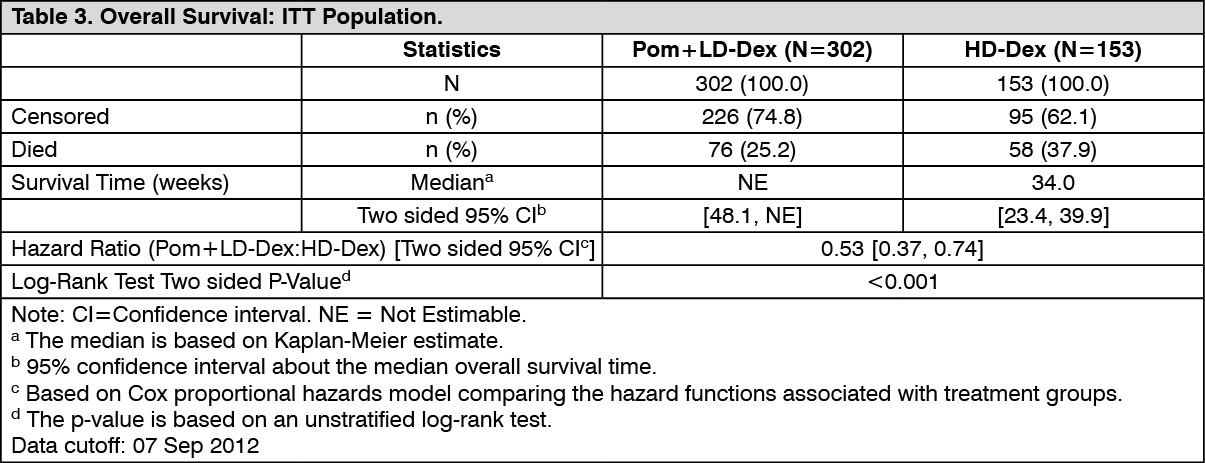
Click on icon to see table/diagram/imageOverall Survival was the key secondary study endpoint. A total of 226 (74.8%) of the Pom+LD-Dex patients and 95 (62.1%) of the HD-Dex patients were alive as of the cutoff date (07 Sep 2012). Median OS time from Kaplan-Meier estimates has not been reached for the Pom+LD-Dex, but would be expected to be at least 48 weeks, which is the lower boundary of the 95% CI. Median OS time for the HD-Dex arm was 34 weeks (95% CI: 23.4, 39.9). The 1-year event free rate was 52.6% (±5.72%) for the Pom+LD-Dex arm and 28.4% (±7.51%) for the HD-Dex arm. The difference in OS between the two treatment arms was statistically significant (p<0.001).
Overall survival is summarised in Table 3 for the ITT population. Kaplan-Meier curve for OS for the ITT population is provided in Figure 3.
Based on the results of both PFS and OS endpoints, the Data Monitoring Committee established for this study recommended that the study be completed and patients in the HD-Dex arm be crossed over to the Pom+LD-Dex arm. (See Table 3 and Figure 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image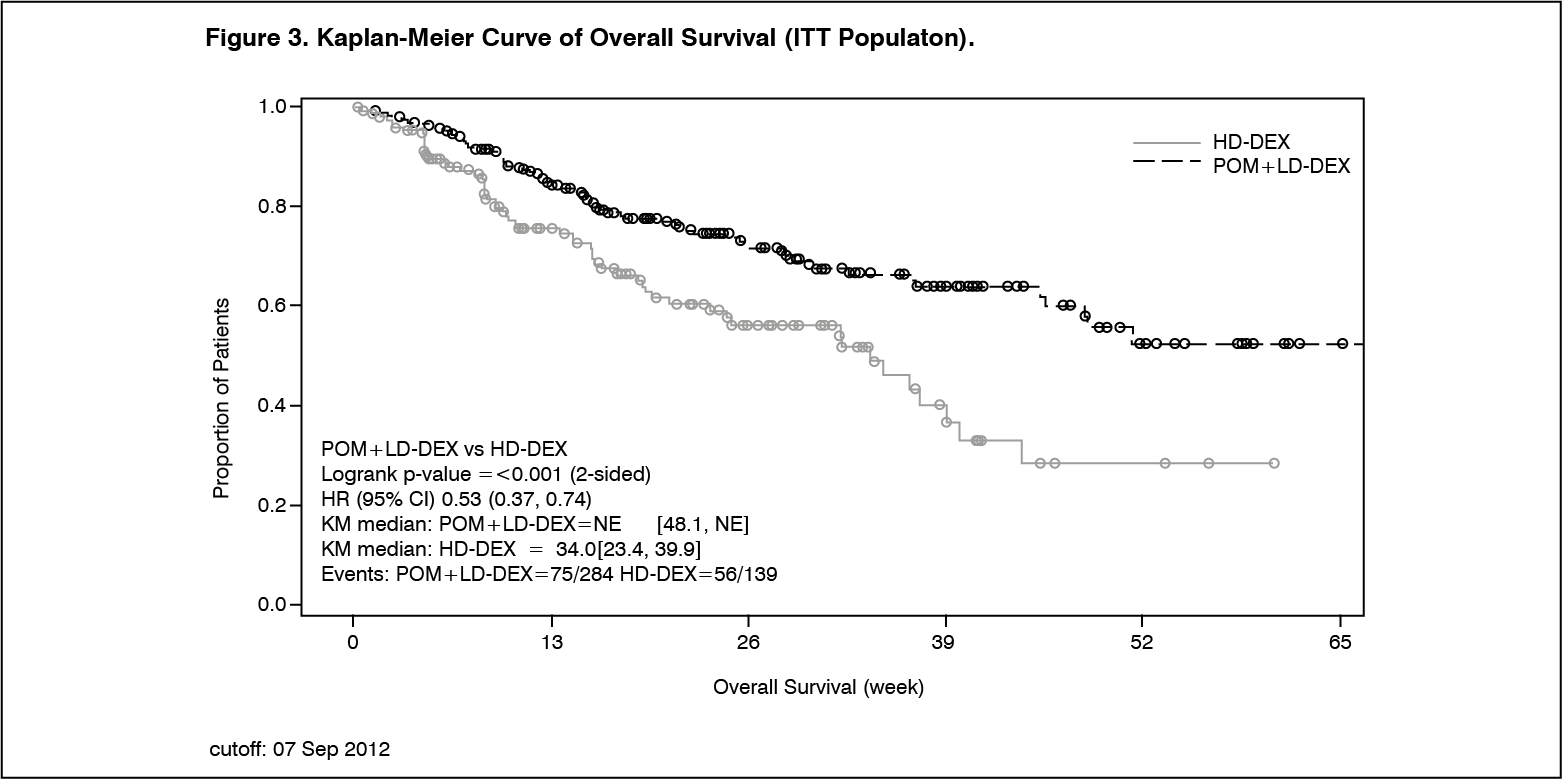 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Absorption: Pomalidomide is absorbed with a maximum plasma concentration (Cmax) occurring between 2 and 3 hours and is at least 73% absorbed following administration of single oral dose. The systemic exposure (AUC) of pomalidomide increases in an approximately linear and dose proportional manner. Following multiple doses, pomalidomide has an accumulation ratio of 27 to 31% on AUC.
Coadministration with a high-fat and high-calorie meal slows the rate of absorption, decreasing mean plasma Cmax by approximately 27%, but has minimal effect on the overall extent of absorption with an 8% decrease in mean AUC. Therefore pomalidomide can be administered without regard to food intake.
Distribution: Pomalidomide has a mean apparent volume of distribution (Vd/F) between 62 and 138 L at steady state. Pomalidomide is distributed in semen of healthy subjects at a concentration of approximately 67% of plasma level at 4 hours post-dose (approximately Tmax) after 4 days of once daily dosing at 2 mg. In vitro binding of pomalidomide enantiomers to proteins in human plasma ranges from 12% to 44% and is not concentration dependent.
Biotransformation: Pomalidomide is the major circulating component (approximately 70% of plasma radioactivity) in vivo in healthy subjects who received a single oral dose of [14C]-pomalidomide (2 mg). No metabolites were present at >10% relative to parent or total radioactivity in plasma.
The predominant metabolic pathways of excreted radioactivity are hydroxylation with subsequent glucuronidation, or hydrolysis. In vitro, CYP1A2 and CYP3A4 were identified as the primary enzymes involved in the CYP-mediated hydroxylation of pomalidomide, with additional minor contributions from CYP2C19 and CYP2D6. Pomalidomide is also a substrate of P-glycoprotein in vitro. Co-administration of pomalidomide with the strong CYP3A4/5 and P-gp inhibitor ketoconazole, or the strong CYP3A4/5 inducer carbamazepine, had no clinically relevant effect on exposure to pomalidomide. Co-administration of the strong CYP1A2 inhibitor fluvoxamine with pomalidomide in the presence of ketoconazole, increased mean exposure to pomalidomide by 107% with a 90% confidence interval [91% to 124%] compared to pomalidomide plus ketoconazole. In a second study to evaluate the contribution of a CYP1A2 inhibitor alone to metabolism changes, co-administration of fluvoxamine alone with pomalidomide increased mean exposure to pomalidomide by 125% with a 90% confidence interval [98% to 157%] compared to pomalidomide alone. If strong inhibitors of CYP1A2 (e.g. ciprofloxacin, enoxacin and fluvoxamine) are co-administered with pomalidomide, reduce the dose of pomalidomide to 50%.
Administration of pomalidomide in smokers, with smoking tobacco known to induce the CYP1A2 isoform, had no clinically relevant effect on exposure to pomalidomide compared to that exposure to pomalidomide observed in non-smokers.
Based on in vitro data, pomalidomide is not an inhibitor or inducer of cytochrome P-450 isoenzymes, and does not inhibit any drug transporters that were studied. Clinically relevant drug-drug interactions are not anticipated when pomalidomide is coadministered with substrates of these pathways.
Elimination: Pomalidomide is eliminated with a median plasma half-life of approximately 9.5 hours in healthy subjects and approximately 7.5 hours in patients with multiple myeloma. Pomalidomide has a mean total body clearance (CL/F) of approximately 7-10 L/hr.
Following a single oral administration of [14C]-pomalidomide (2 mg) to healthy subjects, approximately 73% and 15% of the radioactive dose was eliminated in urine and faeces, respectively, with approximately 2% and 8% of the dosed radiocarbon eliminated as pomalidomide in urine and faeces.
Pomalidomide is extensively metabolised prior to excretion, with the resulting metabolites eliminated primarily in the urine. The 3 predominant metabolites in urine (formed via hydrolysis or hydroxylation with subsequent glucuronidation) account for approximately 23%, 17%, and 12%, respectively, of the dose in the urine.
CYP dependent metabolites account for approximately 43% of the total excreted radioactivity, while non-CYP dependent hydrolytic metabolites account for 25%, and excretion of unchanged pomalidomide accounted for 10% (2% in urine and 8% in faeces).
Population Pharmacokinetics (PK): Based on population PK analysis using a two-compartment model, healthy subjects and MM patients had comparable apparent clearance (CL/F) and apparent central volume of distribution (V2/F). In peripheral tissues, pomalidomide was preferentially taken up by tumors with apparent peripheral distribution clearance (Q/F) and apparent peripheral volume of distribution (V3/F) 3.7-fold and 8-fold higher, respectively, than that of healthy subjects.
Paediatric population: No data are available on administration of pomalidomide to paediatric patients (<18 years of age).
Elderly: Based on population pharmacokinetic analyses in healthy subjects and multiple myeloma patients, no significant influence of age (19-83 years) on oral clearance of pomalidomide was observed. In clinical studies, no dosage adjustment was required in elderly (>65 years) patients exposed to pomalidomide (see Dosage & Administration).
Renal impairment: Population pharmacokinetic analyses showed that the pomalidomide pharmacokinetic parameters were not remarkably affected in renally impaired patients (defined by creatinine clearance or estimated glomerular filtration rate [eGFR]) compared to patients with normal renal function (CrCl ≥60 mL/minute). Mean normalised AUC exposure to pomalidomide was 98.2% with a 90% confidence interval [77.4% to 120.6%] in moderate renal impairment patients (eGFR ≥30 to ≤45 mL/minute/1.73 m2) compared to patients with normal renal function. Mean normalized AUC exposure to pomalidomide was 100.2% with a 90% confidence interval [79.7% to 127.0%] in severe renal impairment patients not requiring dialysis (CrCl <30 or eGFR <30 mL/minute/1.73 m2) compared to patients with normal renal function. Mean normalized AUC exposure to pomalidomide increased by 35.8% with a 90% CI [7.5% to 70.0%] in severe renal impairment patients requiring dialysis (CrCl <30 mL/minute requiring dialysis) compared to patients with normal renal function. The mean changes in exposure to pomalidomide in each of these renal impairment groups are not of a magnitude that require dosage adjustments.
Hepatic impairment: The pharmacokinetic parameters were modestly changed in hepatically impaired patients (defined by Child-Pugh criteria) compared to healthy subjects. Mean exposure to pomalidomide increased by 51% with a 90% confidence interval [9% to 110%] in mildly hepatically impaired patients compared to healthy subjects. Mean exposure to pomalidomide increased by 58% with a 90% confidence interval [13% to 119%] in moderately hepatically impaired patients compared to healthy subjects. Mean exposure to pomalidomide increased by 72% with a 90% confidence interval [24% to 138%] in severely hepatically impaired patients compared to healthy subjects. The mean increases in exposure to pomalidomide in each of these impairment groups are not of a magnitude for which adjustments in schedule or dose are required (see Dosage & Administration).
Toxicology: Preclinical safety data: Repeat-dose toxicology studies: In rats, chronic administration of pomalidomide at doses of 50, 250, and 1000 mg/kg/day for 6 months was well tolerated. No adverse findings were noted up to 1000 mg/kg/day (175-fold exposure ratio relative to a 4 mg clinical dose).
In monkeys, pomalidomide was evaluated in repeat-dose studies of up to 9 months in duration. In these studies, monkeys exhibited greater sensitivity to pomalidomide effects than rats. The primary toxicities observed in monkeys were associated with the haematopoietic/lymphoreticular systems. In the 9-month study in monkeys with doses of 0.05, 0.1, and 1 mg/kg/day, morbidity and early euthanasia of 6 animals were observed at the dose of 1 mg/kg/day and were attributed to immunosuppressive effects (staphylococcal infection, decreased peripheral blood lymphocytes, chronic inflammation of the large intestine, histologic lymphoid depletion, and hypocellularity of bone marrow) at high exposures of pomalidomide (15-fold exposure ratio relative to a 4 mg clinical dose). These immunosuppressive effects resulted in early euthanasia of 4 monkeys due to poor health condition (watery stool, inappetence, reduced food intake, and weight loss); histopathologic evaluation of these animals showed chronic inflammation of the large intestine and villous atrophy of the small intestine. Staphylococcal infection was observed in 4 monkeys; 3 of these animals responded to antibiotic treatment and 1 died without treatment. In addition, findings consistent with acute myelogenous leukemia led to euthanasia of 1 monkey; clinical observations and clinical pathology and/or bone marrow alterations observed in this animal were consistent with immunosuppression. Minimal or mild bile duct proliferation with associated increases in ALP and GGT were also observed at 1 mg/kg/day. Evaluation of recovery animals indicated that all treatment-related findings were reversible after 8 weeks of dosing cessation, except for proliferation of intrahepatic bile ducts observed in 1 animal in the 1 mg/kg/day group. The No Observed Adverse Effect Level (NOAEL) was 0.1 mg/kg/day (0.5-fold exposure ratio relative to a 4 mg clinical dose).
Genotoxicity/carcinogenicity: Pomalidomide was not mutagenic in bacterial and mammalian mutation assays, and did not induce chromosomal aberrations in human peripheral blood lymphocytes or micronuclei formation in polychromatic erythrocytes in bone marrow of rats administered doses up to 2000 mg/kg/day.
Carcinogenicity studies have not been conducted.
Fertility and early embryonic development: In a fertility and early embryonic development study in rats, pomalidomide was administered to males and females at dosages of 25, 250, and 1000 mg/kg/day. Uterine examination on Gestation Day 13 showed a decrease in mean number of viable embryos and an increase in postimplantation loss at all dosage levels. Therefore, the NOAEL for these observed effects was <25 mg/kg/day (AUC24h was 39960 ng·h/mL (nanogram·hour/millilitres) at this lowest dose tested, and the exposure ratio was 99-fold relative to a 4 mg clinical dose). When treated males on this study were mated with untreated females, all uterine parameters were comparable to the controls. Based on these results, the observed effects were attributed to the treatment of females.
Embryo-foetal development: Pomalidomide was found to be teratogenic in both rats and rabbits when administered during the period of major organogenesis. In the rat embryofoetal developmental toxicity study, malformations of absence of urinary bladder, absence of thyroid gland, and fusion and misalignment of lumbar and thoracic vertebral elements (central and/or neural arches) were observed at all dosage levels (25, 250, and 1000 mg/kg/day).
There was no maternal toxicity observed in this study. Therefore, the maternal NOAEL was 1000 mg/kg/day, and the NOAEL for developmental toxicity was <25 mg/kg/day (AUC24h was 34340 ng·h/mL on Gestation Day 17 at this lowest dose tested, and the exposure ratio was 85-fold relative to a 4 mg clinical dose). In rabbits, pomalidomide at dosages ranging from 10 to 250 mg/kg produced embryo-foetal developmental malformations. Increased cardiac anomalies were seen at all doses with significant increases at 250 mg/kg/day. At 100 and 250 mg/kg/day, there were slight increases in post-implantation loss and slight decreases in fetal body weights. At 250 mg/kg/day, fetal malformations included limb anomalies (flexed and/or rotated fore- and/or hindlimbs, unattached or absent digit) and associated skeletal malformations (not ossified metacarpal, misaligned phalanx and metacarpal, absent digit, not ossified phalanx, and short not ossified or bent tibia); moderate dilation of the lateral ventricle in the brain; abnormal placement of the right subclavian artery; absent intermediate lobe in the lungs; low-set kidney; altered liver morphology; incompletely or not ossified pelvis; an increased average for supernumerary thoracic ribs and a reduced average for ossified tarsals. Slight reduction in maternal body weight gain, significant reduction in triglycerides, and significant decrease in absolute and relative spleen weights were observed at 100 and 250 mg/kg/day. The maternal NOAEL was 10 mg/kg/day, and the developmental NOAEL was <10 mg/kg/day (AUC24h was 418 ng·h/mL on Gestation Day 19 at this lowest dose tested, which was similar to that obtained from a 4 mg clinical dose).