


 Sign Out
Sign Out

Pharmacology: Pharmacodynamics: Lamivudine is a potent, selective inhibitor of HIV-1 and HIV-2 replication in vitro. It is also active against zidovudine-resistant clinical isolates of HIV. Lamivudine is metabolised intracellularly to the 5'-triphosphate, the active moiety, which has an intra-cellular half-life of 16 to 19 h. Lamivudine 5'-triphosphate is a weak inhibitor of the RNA and DNA dependant activities of HIV reverse transcriptase, its main mode of action is as a chain terminator of HIV reverse transcription. No antagonistic effects in vitro were seen with lamivudine and other anti retrovirals (tested agents: abacavir, didanosine, nevirapine, zalcitabine, and zidovudine).
Lamivudine does not interfere with cellular deoxynucleotide metabolism and has little effect on mammalian cell and mitochondrial DNA content.
In vitro, lamivudine demonstrates low cytotoxicity to peripheral blood lymphocytes, to established lymphocyte and monocyte-macrophage cell lines, and to a variety of bone marrow progenitor cells in vitro. Lamivudine therefore has, in vitro, a high therapeutic index.
HIV-1 resistance to lamivudine involves the development of a M184V amino acid change close to the active site of the viral reverse transcriptase (RT). This variant arises both in vitro and in HIV-1 infected patients treated with lamivudine-containing antiretroviral therapy. M184V mutants display greatly reduced susceptibility to lamivudine and show diminished viral replicative capacity in vitro. In vitro studies indicate that zidovudine-resistant virus isolates can become zidovudine sensitive when they simultaneously acquire resistance to lamivudine. The clinical relevance of such findings remains, however, not well defined.
Cross-resistance conferred by the M184V RT is limited within the nucleoside inhibitor class of antiretroviral agents. Zidovudine and stavudine maintain their antiretroviral activities against lamivudine-resistant HIV-1. Abacavir maintains its antiretroviral activities against lamivudine-resistant HIV-1 harbouring only the M184V mutation. The M184V RT mutant shows a less than 4-fold decrease in susceptibility to didanosine and zalcitabine; the clinical significance of these findings is unknown. In vitro susceptibility testing has not been standardised and results may vary according to methodological factors.
In clinical trials, lamivudine in combination with zidovudine has been shown to reduce HIV-1 viral load and to increase CD4 cell count. Clinical end-point data indicate that lamivudine in combination with zidovudine alone or in combination with zidovudine containing treatment regimens results in a significant reduction in the risk of disease progression and mortality.
Reduced in vitro sensitivity to lamivudine has been reported for HIV isolates from patients who have received 3TC therapy.
Evidence from clinical studies show that lamivudine plus zidovudine delays the emergence of zidovudine-resistant isolates in individuals with no prior anti-retroviral therapy.
Lamivudine has been widely used as a component of antiretroviral combination therapy with other antiretroviral agents of the same class (nucleoside reverse transcriptase inhibitors) or different classes (protease inhibitors, non-nucleoside reverse transcriptase inhibitors).
Clinical trial evidence from paediatric patients receiving 3TC with other antiretroviral drugs (abacavir, nevirapine/efavirenz or zidovudine) has shown that the resistance profile observed in paediatric patients is similar to that observed in adults, in terms of the genotypic substitutions detected and their relative frequency.
Children receiving 3TC oral solution concomitantly with other antiretroviral oral solutions in clinical trials developed viral resistance more frequently than children receiving tablets (see Pharmacology: Pharmacokinetics and Pharmacodynamics: Clinical Studies under Actions).
Multiple drug antiretroviral therapy containing lamivudine has been shown to be effective in antiretrovirally-naive patients as well as in patients presenting with viruses containing the M184V mutations.
The relationship between in vitro susceptibility of HIV to lamivudine and the clinical response to therapy remain under investigation.
Post-exposure prophylaxis (PEP): Internationally recognised guidelines (Centre for Disease Control and Prevention - June 1998), recommend that in the event of accidental exposure to HIV infected blood e.g. from a needlestick injury, a combination of zidovudine and lamivudine should be administered promptly (within 1 to 2 h). In cases of higher risk of infection a protease inhibitor should be included in the regimen. It is recommended that antiretroviral prophylaxis be continued for four weeks. No controlled clinical studies have been carried out in post-exposure prophylaxis and supporting data is limited. Seroconversion may still occur despite prompt treatment with antiretroviral agents.
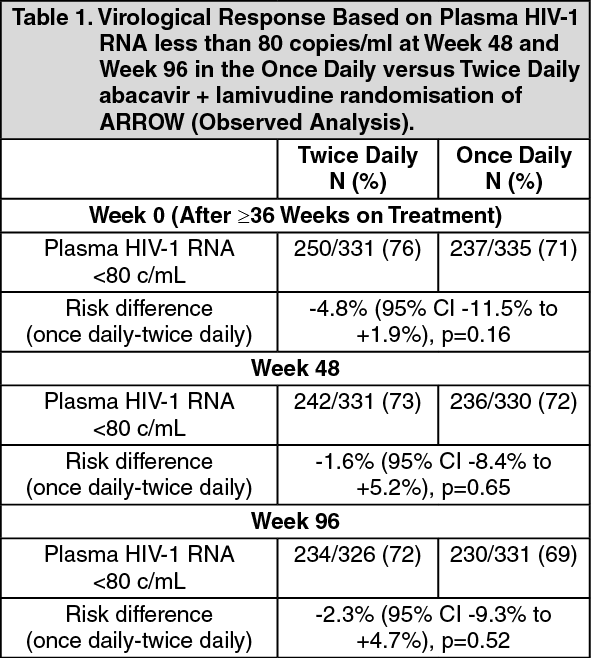
Clinical Studies: A randomised comparison of a regimen including once daily vs twice daily dosing of abacavir and lamivudine was undertaken within a randomised, multicentre, controlled study of HIV-infected, paediatric patients. 1206 paediatric patients aged 3 months to 17 years enrolled in the ARROW Trial (COL105677) and were dosed according to the weight - band dosing recommendations in the World Health Organisation treatment guidelines (Antiretroviral therapy of HIV infection in infants and children, 2006). After 36 weeks on a regimen including twice daily abacavir and lamivudine, 669 eligible subjects were randomised to either continue twice daily dosing or switch to once daily abacavir and lamivudine for at least 96 weeks. The results are summarised in the table as follows: (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe abacavir/lamivudine once daily dosing group was demonstrated to be non-inferior to the twice daily group according to the pre-specified non-inferiority margin of -12%, for the primary endpoint of <80 c/mL at Week 48 as well as at Week 96 (secondary endpoint) and all other thresholds tested (<200c/mL, <400c/mL, <1000c/mL), which all fell well within this non-inferiority margin. Subgroup analyses testing for heterogeneity of once vs twice daily demonstrated no significant effect of sex, age, or viral load at randomisation. Conclusions supported non-inferiority regardless of analysis method.
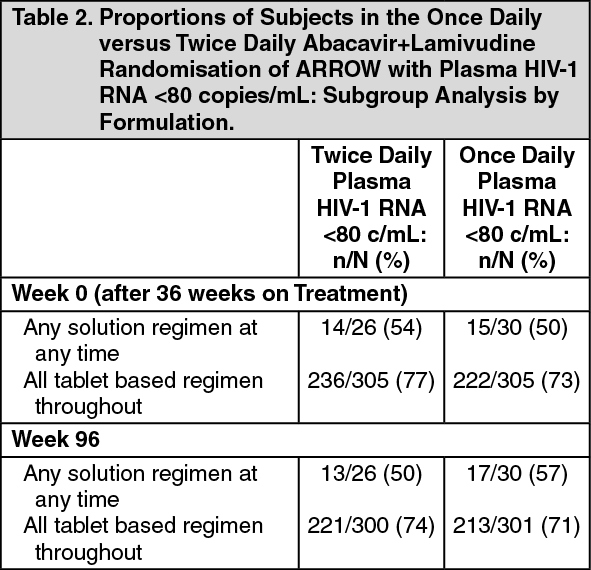
At the time of randomization to once daily vs twice daily dosing (Week 0), those patients who had received tablet formulations had a higher rate of viral load suppression than those who had received any solution formulations at any time. These differences were observed in each different age group studied. This difference in suppression rates between tablets and solutions remained through Week 96 with once daily dosing. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageGenotypic resistance analyses were conducted on samples with plasma HIV-1 RNA >1000 copies/ml. More cases of resistance were detected among patients who had received 3TC solution, in combination with other antiretroviral solutions, compared with those who received similar doses tablet formulation. This is consistent with the lower rates of antiviral suppression observed in these patients.
The Antiretroviral Pregnancy Registry has received reports of over 11,000 exposures to lamivudine during pregnancy resulting in live birth. These consist of over 4,200 exposures during the first trimester, over 6, 900 exposures during the second/third trimester and included 135 and 198 birth defects respectively. The prevalence (95% CI) of defects in the first trimester was 3.2% (2.6, 3.7%) and in the second/third trimester, 2.8% (2.4, 3.2%). Among pregnant women in the reference population, the background rate of birth defects is 2.7%. The Antiretroviral Pregnancy Registry does not show an increased risk of major birth defects for lamivudine compared to the background rate.
Pharmacokinetics: Absorption: Lamivudine is well absorbed from the gastrointestinal tract, and the bioavailability of oral lamivudine in adults is normally between 80 and 85%. Following oral administration, the mean time (tmax) to maximal serum concentrations (Cmax) is about an hour. At therapeutic dose levels i.e. 4 mg/kg/day (as two 12-hourly doses), Cmax is in the order of 1 to 1.9 micrograms/ml.
Co-administration of lamivudine with food resulted in a delay of tmax and a lower Cmax (decreased by up to 47%). However, the extent (based on the AUC) of lamivudine absorbed was not influenced. No dose adjustment is needed when co-administered with food.
Administration of crushed tablets with a small amount of semi-solid food or liquid would not be expected to have an impact on the pharmaceutical quality, and would therefore not be expected to alter the clinical effect. This conclusion is based on the physicochemical and pharmacokinetic characteristics of the active ingredient and the in vitro dissolution behaviour of lamivudine tablets in water, assuming that the patient crushes and transfers 100% of the tablet and ingests immediately.
FC tablet: Administration of two 150 mg tablets is bioequivalent to administration of one 300 mg tablet with respect to AUC∞, Cmax, and tmax. Administration of tablets is bioequivalent to oral solution with respect to AUC∞ and Cmax in adults. Absorption differences have been observed between adult and pediatric populations (see special patient populations/children as follows).
Distribution: From i.v. studies, the mean volume of distribution is 1.3 l/kg and the mean terminal half-life of elimination is 5 to 7 h.
Lamivudine exhibits linear pharmacokinetics over the therapeutic dose range and displays low plasma protein binding to albumin.
Limited data shows lamivudine penetrates the central nervous system and reaches the cerebro-spinal fluid (CSF). The mean lamivudine CSF/serum concentration ratio 2 to 4 h after oral administration was approximately 0.12. The true extent of penetration or relationship with any clinical efficacy is unknown.
Metabolism and Elimination: Lamivudine mean systemic clearance is approximately 0.32 l/h/kg, with predominantly renal clearance (greater than 70%) via the organic cationic transport system, and little (less than 10%) hepatic metabolism.
The active moiety, intracellular lamivudine triphosphate, has a prolonged terminal half-life in the cell (16 to 19 h) compared to the plasma lamivudine half-life (5 to 7 h). In 60 healthy adult volunteers, lamivudine 300 mg once daily has been demonstrated to be pharmacokinetically equivalent at steady-state to lamivudine 150 mg twice daily with respect to intracellular triphosphate AUC24 and Cmax.
The likelihood of adverse interactions between lamivudine and other medicinal products is low due to limited metabolism and plasma protein binding and almost complete renal elimination of unchanged lamivudine.
Special Patient Populations: Children: The absolute bioavailability of lamivudine (approximately 58 to 66%) was lower and more variable in paediatric patients under 12 years of age. In children, administration of tablets given concomitantly with other antiretroviral tablets delivered higher plasma lamivudine AUC∞ and Cmax than oral solution given concomitantly with other antiretroviral oral solutions. Children receiving 3TC oral solution according to the recommended dosage regimen achieve plasma lamivudine exposure within the range of values observed in adults. Children receiving 3TC oral tablets according to the recommended dosage regimen achieve higher plasma lamivudine exposure than children receiving oral solution because higher mg/kg doses are administered with the tablet formulation and the tablet formulation has higher bioavailability (see Dosage & Administration). Paediatric pharmacokinetics studies with both oral solution and tablet formulations have demonstrated that once daily dosing provides equivalent AUC0-24 to twice daily dosing of the same total daily dose.
There are limited pharmacokinetic data for patients less than three months of age. In neonates one week of age, lamivudine oral clearance was reduced when compared to paediatric patients and is likely to be due to immature renal function and variable absorption. Therefore to achieve similar adult and paediatric exposure, the recommended dose for neonates is 2 mg/kg twice a day. However there is no data available in neonates older than one week old.
Elderly: No pharmacokinetic data are available in patients over 65 years of age.
Renal impairment: Lamivudine plasma concentrations (AUC) are increased in patients with renal dysfunction due to decreased clearance. The dosage should therefore be reduced for patients with a creatinine clearance of less than 50 ml/min (see Dosage & Administration).
Hepatic impairment: Data obtained in patients with moderate to severe hepatic impairment show that lamivudine pharmacokinetics are not significantly affected by hepatic dysfunction.
Pregnancy: The pharmacokinetics of lamivudine are similar to that of non-pregnant adults. In humans, consistent with passive transmission of lamivudine across the placenta, lamivudine concentrations in infant serum at birth were similar to those in maternal and cord serum at delivery.
Toxicology: Pre-clinical Safety Data: Carcinogenesis, mutagenesis: Lamivudine was not mutagenic in bacterial tests but, like many nucleoside analogues, showed activity in an in vitro cytogenetic assay and the mouse lymphoma assay. Lamivudine was not genotoxic in vivo at doses that gave plasma concentrations around 40 to 50 times higher than the anticipated clinical plasma levels. As the in vitro mutagenic activity of lamivudine could not be confirmed in in vivo tests, it is concluded that 3TC should not represent a genotoxic hazard to patients undergoing treatment.
The results of long term oral carcinogenicity studies with lamivudine in rats and mice did not show any carcinogenic potential.
Reproductive toxicology: Reproductive studies in animals have not shown evidence of teratogenicity, and showed no effect on male or female fertility. Lamivudine produced small increases in early embryonic loss when administered to pregnant rabbits, at exposure levels comparable to those achieved in man. However, there was no evidence of embryonic loss in rats at exposure levels of approximately 35 times the clinical exposure (based on Cmax).
Animal toxicology: Administration of lamivudine in animal toxicity studies at very high doses was not associated with any major organ toxicity. Reductions of erythrocyte and neutrophil counts were identified as the effects most likely to be of clinical relevance.