


 Sign Out
Sign Out
Pharmacology: Pharmacodynamics: Mechanism of Action: KRABEVA (bevacizumab) is a recombinant humanised monoclonal antibody that selectively binds to and neutralises the biologic activity of human vascular endothelial growth factor (VEGF). Bevacizumab contains human framework regions with antigen binding regions of a humanised murine antibody that binds to VEGF. Bevacizumab is produced by recombinant DNA technology in a Chinese Hamster ovary mammalian cell expression system and is purified by a process that includes specific viral inactivation and removal steps. Bevacizumab consists of 214 amino acids and has a molecular weight of approximately 149,000 daltons.
Bevacizumab inhibits the binding of VEGF to its receptors, Flt-1 and KDR, on the surface of endothelial cells. Neutralising the biologic activity of VEGF reduces the vascularisation of tumours, thereby inhibiting tumour growth. Administration of bevacizumab or its parental murine antibody to xenotransplant models of cancer in nude mice resulted in extensive antitumour activity in human cancers, including colon, breast, pancreas and prostate. Metastatic disease progression was inhibited and microvascular permeability was reduced.
Clinical/Efficacy Studies: Efficacy data of KRABEVA: Study MYL-1402O-3001: A total of 671 patients were randomized with 337 patients to the KRABEVA arm and 334 patients to the Avastin arm. The primary efficacy endpoint of this Phase III study in NSCLC was the ORR as assessed by an independent review during the first 18 Weeks, according to RECIST 1.1. At Week 18, 217 (64.4%) patients in the KRABEVA arm and 225 (67.4%) in the Avastin arm had their treatment ongoing. The primary efficacy endpoint was based on the ratio of ORRs. The equivalence of both treatments was defined as the two-sided 90% CI for the ratio of ORRs at Week 18 being entirely within the equivalence range of 0.73, 1.36. The ratio of ORR (KRABEVA: Avastin) between the treatment groups was 0.96 (90% CI 0.83, 1.12). As this CI was entirely within the pre-defined equivalence margin, therapeutic equivalence of KRABEVA and Avastin was statistically confirmed.
Immunogenicity Results: A total of 12 (3.6%) and 16 patients (5.0%) were positive for ADA at baseline in the KRABEVA and Avastin arms, respectively. The incidence of baseline ADA positivity in a small proportion of patients has been observed in other bevacizumab studies and may be due to potential cross-reactivity with pre-existing antibodies and highly sensitive nature of the assay. Post-baseline, the number of ADA positive patients were low, declined over time and were comparable between both treatment arms. The ADA titers were found to be comparable in the 2 arms.
Study BM100-CC-03-I-01: Efficacy of KRABEVA and Avastin (both in combination with XELOX chemotherapy) was compared in terms of best overall response rate (BORR) over 18 weeks in a Phase III study in patients with mCRC. BORR was determined in the PP as well as the ITT populations where objective response was defined as a CR or PR. PR was observed in 25 patients in the KRABEVA arm and 32 patients in the Avastin arm. The ORR observed in the ITT population was 38.24% in the KRABEVA arm and 48.53% in the Avastin arm. No statistically significant difference was observed between the two arms (p=0.2258). The ORR observed in this study is similar to historically observed ORR of 36.6% when bevacizumab is used in combination with chemotherapy.
The PFS rate was similar in both arms at 18 weeks (61.76% in the KRABEVA and 60.29% in the Avastin arm). No statistically significant difference between the two arms was observed for PFS rate (p = 0.8604).
Due to the prominent effect of bevacizumab on disease stabilization; clinical benefit to the patients based on DCR was analyzed and compared between the two arms.DCR is defined as the sum of CR, PR and SD based on the best overall response evaluated as per the RECIST 1.1. The DCR was 91.18% in the KRABEVA arm and 88.24% in the Avastin arm in the ITT population. The analysis of the DCR revealed no notable differences between both the arms for the proportion of patients with disease control (p=0.5399).
All studies performed in every single indication as approved are referring to the studies conducted by AVASTIN.
Metastatic Colorectal Cancer (mCRC): The safety and efficacy of the recommended dose of Bevacizumab (5 mg/kg of body weight every two weeks) in metastatic carcinoma of the colon or rectum were studied in three randomized, active-controlled clinical trials in combination with fluoropyrimidine-based first-line chemotherapy. Bevacizumab was combined with two chemotherapy regimens: AVF2107g: A weekly schedule of irinotecan/bolus 5-fluorouracil/leucovorin (IFL regimen) for a total of 4 weeks of each 6-week cycle.
AVF0780g: In combination with bolus 5-fluorouracil/leucovorin (5-FU/LV) for a total of 6 weeks of each 8-week cycle (Roswell Park regimen).
AVF2192g: In combination with bolus 5-fluorouracil/leucovorin (5-FU/LV) for a total of 6 weeks of each 8 week-cycle (Roswell Park regimen) in patients who were not optimal candidates for first-line irinotecan treatment.
Three additional studies with Bevacizumab have been conducted in mCRC patients: first-line (No16966), second-line with no previous Bevacizumab treatment (E3200), and second-line with previous Bevacizumab treatment following disease progression in first-line (ML18147). In these studies, Bevacizumab was administered at the following dosing regimens, in combination with FOLFOX-4 (5FU/LV/Oxaliplatin), XELOX (Capecitabine/Oxaliplatin) and fluoropyrimidine/irinotecan and fluoropyrimidine/oxaliplatin: NO16966: Bevacizumab 7.5 mg/kg of body weight every 3 weeks in combination with oral capecitabine and intravenous oxaliplatin (XELOX) or Bevacizumab 5mg/kg every 2 weeks in combination with leucovorin plus 5-fluorouracil bolus, followed by 5-fluorouracil infusion, with intravenous oxaliplatin (FOLFOX-4).
E3200: Bevacizumab 10 mg/kg of body weight every 2 weeks in combination with leucovorin and 5-fluorouracil bolus, followed by 5-fluorouracil infusion, with intravenous oxaliplatin (FOLFOX-4) in Bevacizumab naïve patients.
ML18147: Bevacizumab 5.0 mg/kg of body weight every 2 weeks or Bevacizumab 7.5 mg/kg of body weight every 3 weeks in combination with fluoropyrimidine/irinotecan or fluoropyrimidine/oxaliplatin in patients with disease progression following first-line treatment with Bevacizumab. Use of irinotecan- or oxaliplatin-containing regimen was switched depending on first-line usage of either oxaliplatin or irinotecan.
AVF2107g: This was a phase III randomised, double-blind, active-controlled clinical trial evaluating Bevacizumab in combination with IFL as first-line treatment for metastatic carcinoma of the colon or rectum. Eight hundred and thirteen patients were randomised to receive IFL + placebo (Arm 1) or IFL + Bevacizumab (5 mg/kg every 2 weeks, Arm 2). A third group of 110 patients received bolus 5-FU/LV + Bevacizumab (Arm 3). Enrolment in Arm 3 was discontinued, as pre-specified, once safety of Bevacizumab with the IFL regimen was established and considered acceptable.
The primary efficacy parameter of the trial was overall survival. The addition of Bevacizumab to IFL resulted in a statistically significant increase in overall survival, progression-free survival and overall response rate (see Table 1 for details). The clinical benefit of Bevacizumab, as measured by survival, was seen in all pre-specified patient subgroups, including those defined by age, sex, performance status, and location of primary tumour, number of organs involved, and duration of metastatic disease. (See Table 1.)
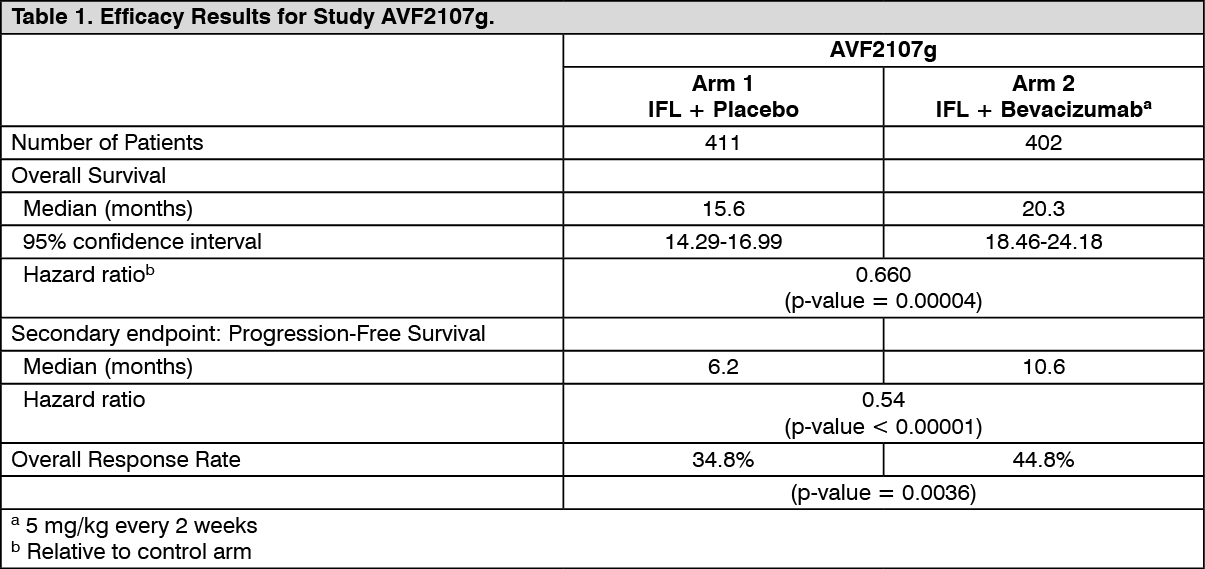 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAmong the 110 patients randomised to Arm 3 (5-FU/LV + Bevacizumab) prior to discontinuation of this arm, the median overall survival was 18.3 months, and the median progression free survival was 8.8 months.
AVF2192g: This was a phase II randomised, double-blind, active-controlled clinical trial investigating Bevacizumab in combination with 5-FU/leucovorin as first-line treatment for metastatic colorectal cancer in patients who were not optimal candidates for first-line irinotecan treatment. One hundred and five patients were randomised to 5-FU/LV + placebo arm and 104 patients randomised to 5-FU/LV + Bevacizumab (5 mg/kg every 2 weeks). All treatments were continued until disease progression.
The addition of Bevacizumab 5 mg/kg every two weeks to 5-FU/LV resulted in higher objective response rates, significantly longer progression-free survival, and a trend in longer survival as, compared with 5-FU/LV chemotherapy alone.
NO16966: This was a phase III randomised, double-blind (for bevacizumab), clinical trial investigating Bevacizumab 7.5 mg/kg in combination with oral capecitabine and i.v. oxaliplatin (XELOX), administered on a 3-weekly schedule; or Bevacizumab 5 mg/kg in combination with leucovorin with 5-fluorouracil bolus, followed by 5-fluorouracil infusional, with i.v. oxaliplatin (FOLFOX-4), administered on a 2-weekly schedule. The study contained two parts: an initial unblinded 2-arm part (Part I) in which patients were randomized to two different treatment groups (XELOX and FOLFOX-4) and a subsequent 2 x 2 factorial 4-arm part (Part II) in which patients were randomized to four treatment groups (XELOX + placebo, FOLFOX-4 + placebo, XELOX + Bevacizumab, FOLFOX-4 + Bevacizumab). In Part II, treatment assignment was double-blind with respect to Bevacizumab.
Approximately 350 patients were randomized into each of the 4 study arms in the Part II of the trial. (See Table 2.)
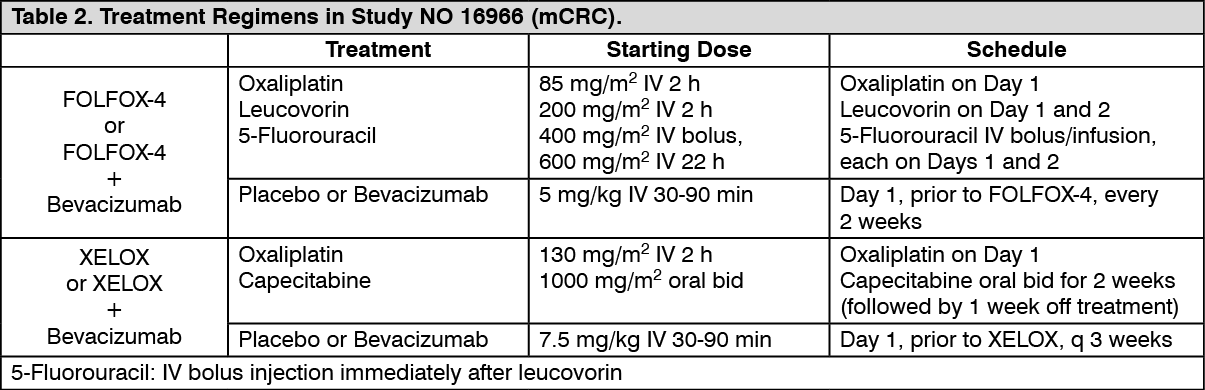 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe primary efficacy parameter of the trial was the duration of progression-free survival. In this study, there were two primary objectives: to show that XELOX was non-inferior to FOLFOX-4 and to show that Bevacizumab in combination with FOLFOX-4 or XELOX chemotherapy was superior to chemotherapy alone. Both coprimary objectives were met: i) Non-inferiority of the XELOX-containing arms compared with the FOLFOX-4-containing arms in the overall comparison was demonstrated in terms of progression-free survival and overall survival in the eligible per-protocol population; ii) Superiority of the Bevacizumab-containing arms versus the chemotherapy alone arms in the overall comparison was demonstrated in terms of progression-free survival in the ITT population (Table 3).
Secondary PFS analyses, based on Independent Review Committee (IRC) - and 'on treatment' - based response assessments, confirmed the significantly superior clinical benefit for patients treated with Bevacizumab (subgroup analyses shown in Table 3), consistent with the statistically significant benefit observed in the pooled analysis. (See Table 3.)
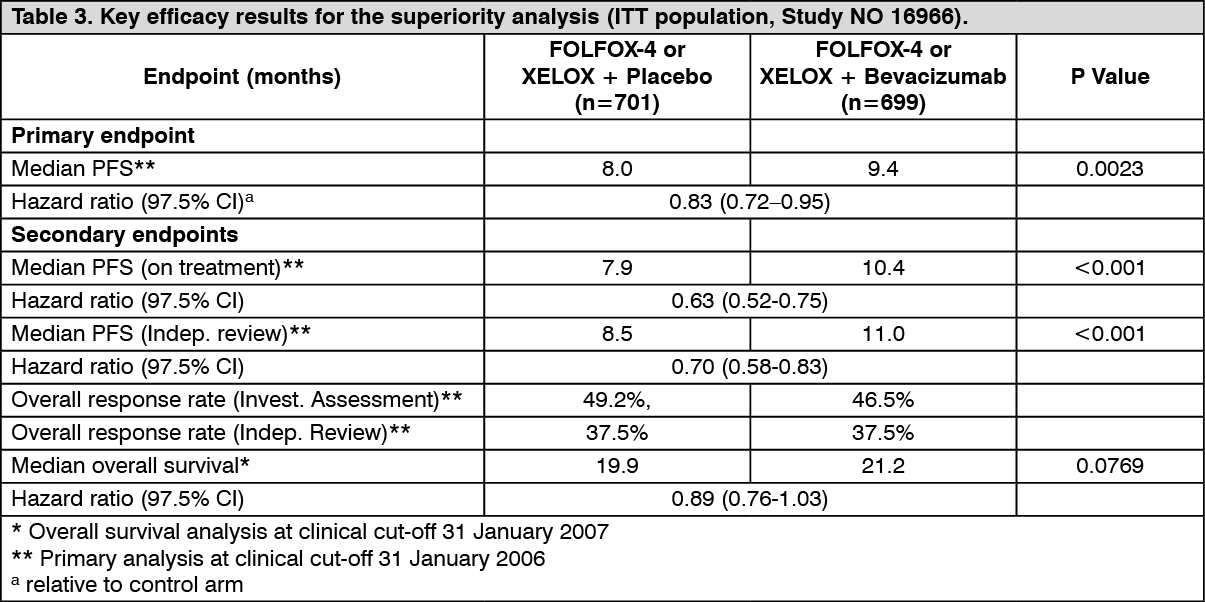 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageECOG E3200: This was a phase III randomised, active-controlled, open-label study investigating Bevacizumab 10 mg/kg in combination with leucovorin with 5-fluorouracil bolus and then 5-fluorouracil infusional, with iv oxaliplatin (FOLFOX-4), administered on a 2-weekly schedule in previously-treated patients (second line) with advanced colorectal cancer. In the chemotherapy arms, the FOLFOX-4 regimen used the same doses and schedule as shown in Table 4 for Study NO16966.
The primary efficacy parameter of the trial was overall survival, defined as the time from randomization to death from any cause. Eight hundred and twenty-nine patients were randomized (292 FOLFOX-4, 293 Bevacizumab + FOLFOX-4 and 244 Bevacizumab monotherapy). The addition of Bevacizumab to FOLFOX-4 resulted in a statistically significant prolongation of survival. Statistically significant improvements in progression-free survival and objective response rate were also observed (see Table 4).
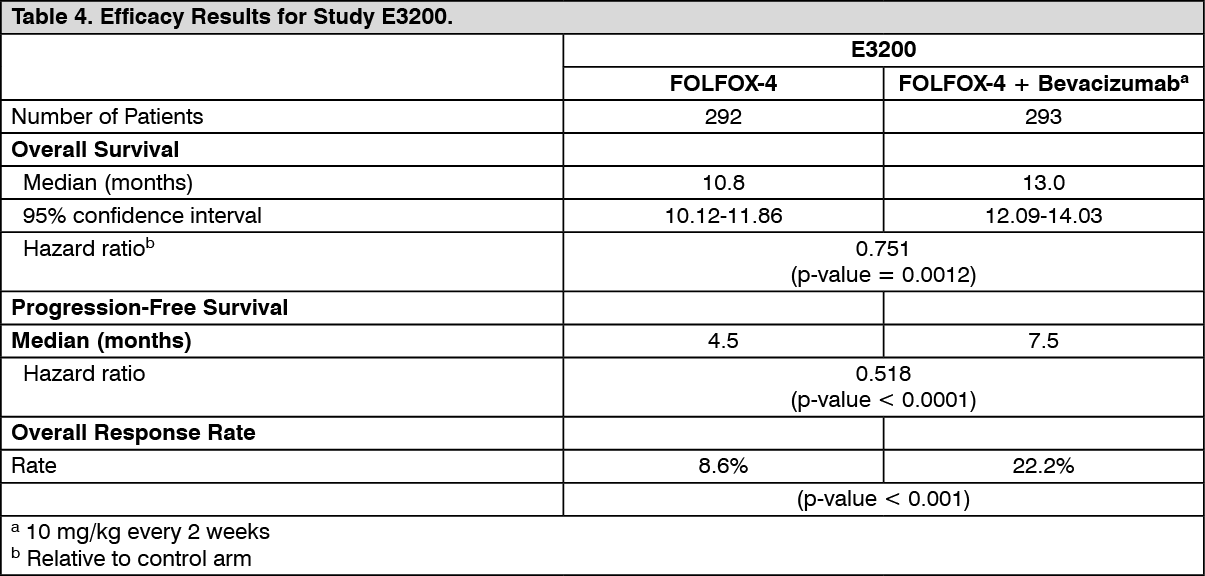 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageNo significant difference was observed in the duration of overall survival between patients who received Bevacizumab monotherapy compared to patients treated with FOLFOX-4.
Progression-free survival and objective response rate were inferior in the Bevacizumab monotherapy arm compared to the FOLFOX-4 arm.
Ml18147: This was a Phase III randomized, controlled, open-label trial investigating Bevacizumab 5.0 mg/kg every 2 weeks or 7.5 mg/kg every 3 weeks in combination with fluoropyrimidine-based chemotherapy versus fluoropyrimidine-based chemotherapy alone in patients with metastatic colorectal cancer who have progressed on a first-line Bevacizumab-containing regimen.
Patients with histologically confirmed mCRC and disease progression were randomized 1:1 within 3 months after discontinuation of Bevacizumab first-line therapy to receive fluoropyrimidine/oxaliplatin or fluoropyrimidine/irinotecan-based chemotherapy (chemotherapy switched depending on first-line chemotherapy) with or without Bevacizumab. Treatment was given until progressive disease or unacceptable toxicity. The primary outcome measure was overall survival (OS) defined as the time from randomization until death from any cause.
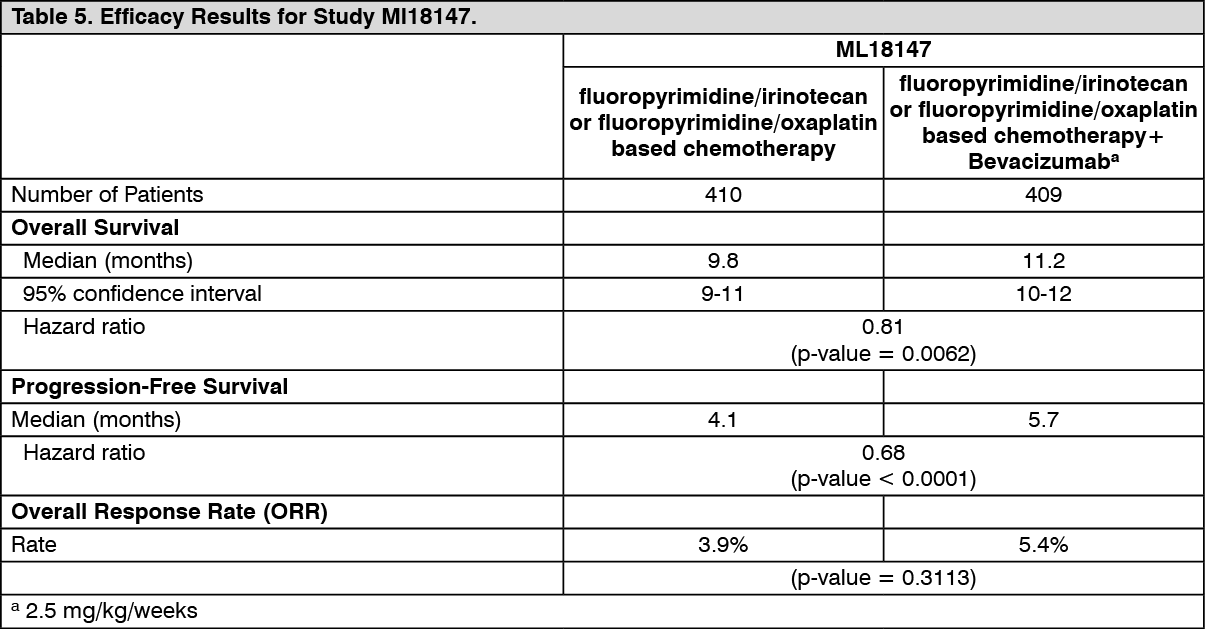
A total of 820 patients were randomized. The addition of Bevacizumab to fluoropyrimidine-based chemotherapy resulted in a statistically significant prolongation of survival in patients with metastatic colorectal cancer who have progressed on a first-line Bevacizumab-containing regimen (ITT = 819) (see Table 5).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageStatistically significant improvements in progression-free survival were also observed.
Objective response rate was low in both treatment arms and did not meet statistical significance.
Adjuvant Colon Cancer (aCC): BO17920: This was a phase III randomized open-label, 3-arm study evaluating the efficacy and safety of Bevacizumab administered at a dose equivalent to 2.5 mg/kg/week on either a 2-weekly schedule in combination with FOLFOX-4, or on a 3-weekly schedule in combination with XELOX versus FOLFOX-4 alone as adjuvant chemotherapy in 3451 patients with high-risk stage II and stage III colon carcinoma.
More relapses and deaths due to disease progression were observed in both Bevacizumab arms compared to the control arm. The primary objective of prolonging disease-free survival (DFS) in patients with stage III colon cancer (n = 2867) by adding Bevacizumab to either chemotherapy regimen was not met. The hazard ratios for DFS were 1.17 (95% CI: 0.98- 1.39) for the FOLFOX-4 + Bevacizumab arm and 1.07 (95% CI: 0.90-1.28) for the XELOX + Bevacizumab arm.
Metastatic Breast Cancer (mBC): ECOG E2100: E2100 was an open-label, randomised, active controlled, multicentre clinical trial evaluating Bevacizumab in combination with paclitaxel for locally recurrent or metastatic breast cancer in patients who had not previously received chemotherapy for locally recurrent and metastatic disease. Prior hormonal therapy for the treatment of metastatic disease was allowed. Adjuvant taxane therapy was allowed only if it was completed at least 12 months prior to study entry.
Patients were randomised to paclitaxel alone (90 mg/m2 IV over 1 hour once weekly for three out of four weeks) or in combination with Bevacizumab (10 mg/kg IV infusion every two weeks). Patients were to continue assigned study treatment until disease progression. In cases where patients discontinued chemotherapy prematurely, treatment with Bevacizumab as a single agent was continued until disease progression. The primary endpoint was progression free survival (PFS), as assessed by investigators. In addition, an independent review of the primary endpoint was also conducted.
Of the 722 patients in the study, the majority of patients (90%) had HER2-negative disease. A small number of patients had HER-2 receptor status that was either unknown (8%) or positive (2%). Patients who were HER-2 positive had either received previous treatment with trastuzumab or were considered unsuitable for trastuzumab. The majority (65%) of patients had received adjuvant chemotherapy including 19% who had prior taxanes and 49% who had prior anthracyclines. The patient characteristics were similar between the study arms.
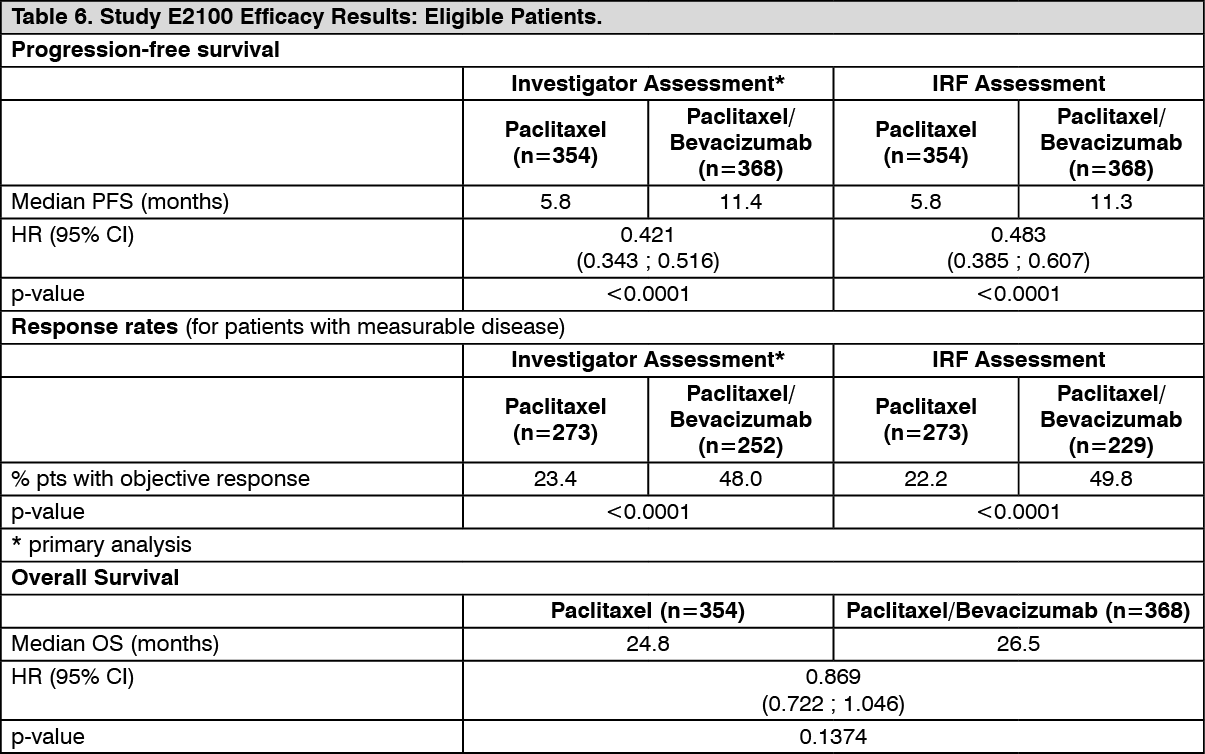
The results of this study are presented in Table 6. (See Table 6.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdvanced, metastatic or recurrent Non-Small Cell Lung Cancer (NSCLC): The safety and efficacy of Bevacizumab in the first-line treatment of patients with non-small cell lung cancer (NSCLC) other than predominantly squamous cell histology, was studied in addition to platinum-based chemotherapy in studies E4599 and BO17704.
E4599: E4599 was an open-label, randomised, active-controlled, multicentre clinical trial evaluating Bevacizumab as first-line treatment of patients with locally advanced, metastatic or recurrent NSCLC other than predominantly squamous cell histology. Patients were randomized to platinum-based chemotherapy (paclitaxel 200 mg/m2 and carboplatin AUC = 6.0, both by IV infusion) (PC) on day 1 of every 3-week cycle for up to 6 cycles or PC in combination with Bevacizumab at a dose of 15 mg/kg IV infusion day 1 of every 3-week cycle. After completion of six cycles of carboplatin-paclitaxel chemotherapy or upon premature discontinuation of chemotherapy, patients on the Bevacizumab + carboplatin-paclitaxel arm continued to receive Bevacizumab as a single agent every 3 weeks until disease progression. 878 patients were randomised to the two arms.
During the study, of the patients who received trial treatment, 32.2% (136/422) of patients received 7-12 administrations of Bevacizumab and 21.1% (89/422) of patients received 13 or more administrations of Bevacizumab.
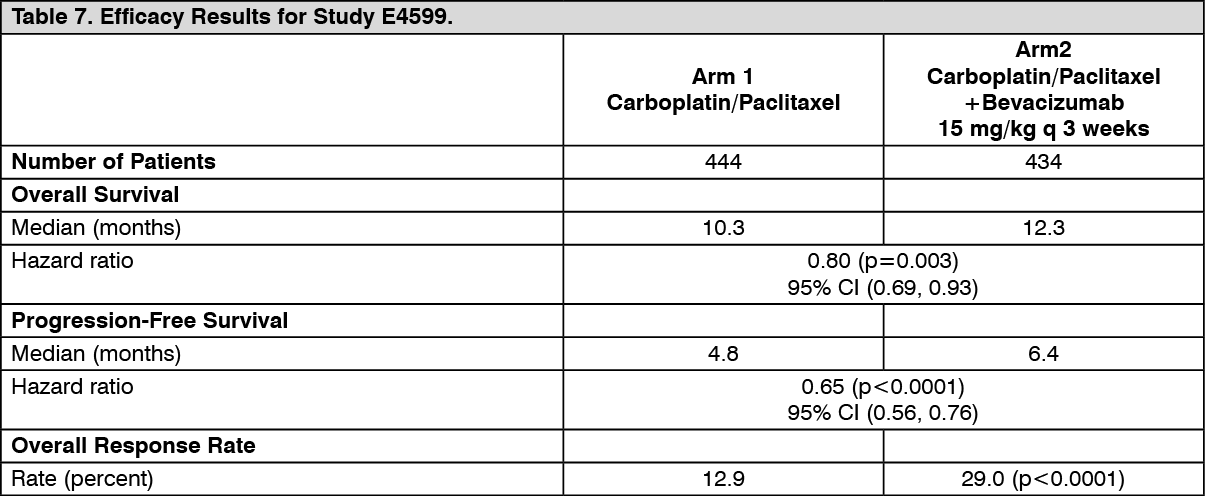
The primary endpoint was duration of survival. Results are presented in Table 7. (See Table 7.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageBO17704: Study BO17704 was a randomised, double-blind phase III study of Bevacizumab in addition to cisplatin and gemcitabine versus placebo, cisplatin and gemcitabine in patients with locally advanced, metastatic or recurrent non-squamous NSCLC, who had not received prior chemotherapy. The primary endpoint was progression free survival, secondary endpoints for the study included the duration of overall survival.
Patients were randomised to platinum-based chemotherapy, cisplatin 80 mg/m2 i.v. infusion on day 1 and gemcitabine 1250 mg/m2 i.v. infusion on days 1 and 8 of every 3-week cycle for up to 6 cycles (CG) with placebo or CG with Bevacizumab at a dose of 7.5 or 15 mg/kg IV infusion day 1 of every 3-week cycle. In the Bevacizumab-containing arms, patients could receive Bevacizumab as a single agent every 3 weeks until disease progression or unacceptable toxicity.
Study results show that 94% (277/296) of eligible patients went on to receive single agent bevacizumab at cycle 7. A high proportion of patients (approximately 62%) went on to receive a variety of non-protocol specified anti-cancer therapies, which may have impacted the analysis of overall survival.
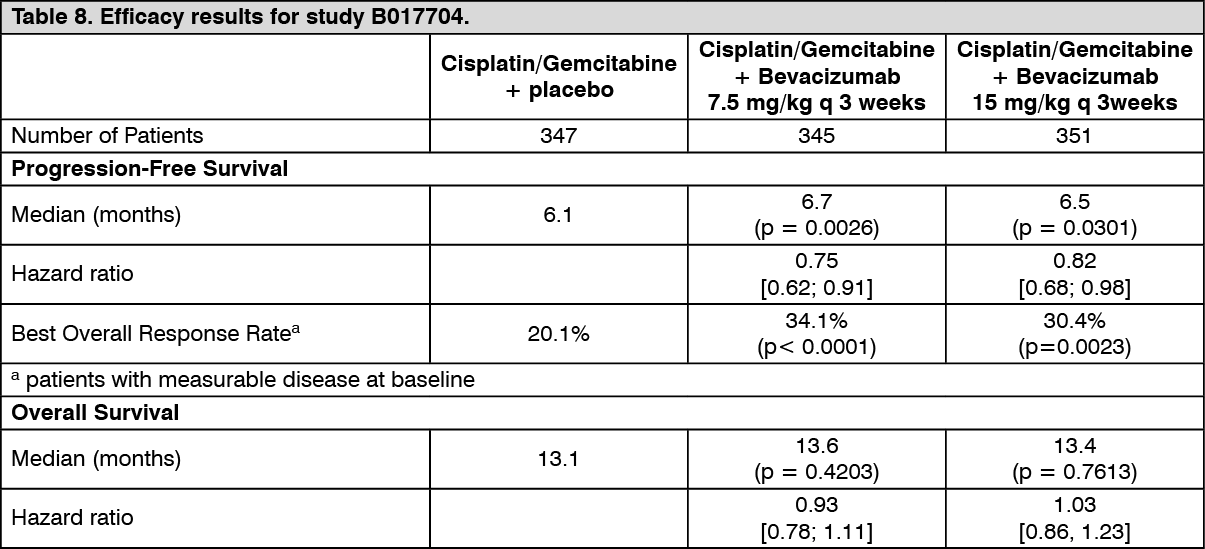
The efficacy results are presented in Table 8. (See Table 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAdvanced and/or metastatic Renal Cell Cancer (mRCC): BO17705: Study BO17705 was a multicenter randomised, double-blind phase III trial conducted to evaluate the efficacy and safety of Bevacizumab in combination with interferon (IFN)-alfa-2a (Roferon) versus IFN-alfa-2a alone as first-line treatment in mRCC. The 649 randomized patients (641 treated) had clear cell mRCC, Karnofsky Performance Status (KPS) of ≥70%, no CNS metastases and adequate organ function. IFN-alfa-2a (x3/week at a recommended dose of 9 MIU) plus Bevacizumab (10 mg/kg q2w) or placebo was given until disease progression. Patients were stratified according to country and Motzer score and the treatment arms were shown to be well balanced for the prognostic factors.
The primary endpoint was overall survival, with secondary endpoints for the study including progression-free survival. The addition of Bevacizumab to IFN-alfa-2a significantly increased PFS and objective tumour response rate. These results have been confirmed through an independent radiological review. However, the increase in the primary endpoint of overall survival by 2 months was not significant (HR = 0.91). A high proportion of patients (approximately 63% IFN/placebo; 55% Bevacizumab/IFN) received a variety of nonspecified, post-protocol anti-cancer therapies, including antineoplastic agents, which may have impacted the analysis of overall survival.
The efficacy results are presented in Table 9. (See Table 9.)
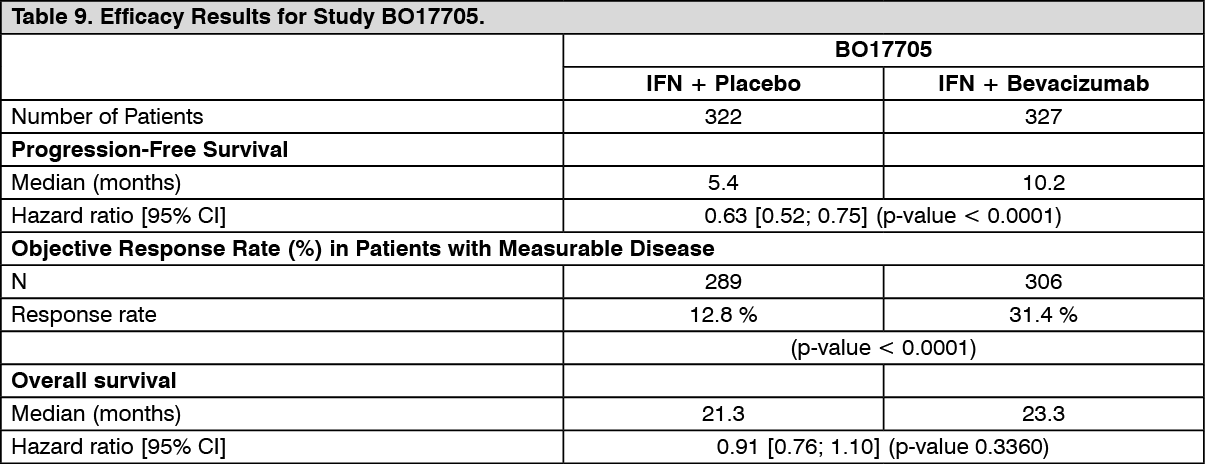 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAn exploratory multivariate Cox regression model using backward selection indicated, that the following baseline prognostic factors were strongly associated with survival independent of treatment: gender, white blood cell count, platelets, body weight loss in the 6 months prior to study entry, number of metastatic sites, sum of longest diameter of target lesions, Motzer score. Adjustment for these baseline factors resulted in a treatment hazard ratio of 0.78 (95% CI [0.63; 0.96], p = 0.0219), indicating a 22% reduction in the risk of death for patients in the Bevacizumab + IFN alfa-2a arm compared to IFN alfa-2a arm.
Ninety-seven (97) patients in the IFN alfa-2a arm and 131 patients in the Bevacizumab arm reduced the dose of IFN alfa-2a from 9 MIU to either 6 or 3 MIU, three times a week as pre-specified in the protocol. Dose-reduction of IFN alfa-2a did not appear to affect the efficacy of the combination of Bevacizumab and IFN alfa-2a, based on PFS event free rates over time, as shown by a sub-group analysis. The 131 patients in the Bevacizumab + IFN alfa-2a arm who reduced and maintained the IFN alfa-2a dose at 6 or 3 MIU during the study, exhibited at 6, 12 and 18 months, PFS event free rates of 73, 52 and 21% respectively, as compared to 61, 43 and 17% in the total population of patients receiving Bevacizumab + IFN alfa-2a.
AVF2938: This was a randomized, double-blind, phase II clinical study investigating Bevacizumab 10 mg/kg in a 2 weekly schedule with the same dose of Bevacizumab in combination with 150 mg daily erlotinib, in patients with metastatic clear cell RCC. A total of 104 patients were randomised to treatment in this study, 53 to Bevacizumab 10 mg/kg q2w plus placebo and 51 to Bevacizumab 10 mg/kg q2w plus erlotinib 150 mg daily. The analysis of the primary endpoint showed no difference between the Bevacizumab + Pl arm and the Bevacizumab + Erl arm (median PFS 8.5 versus 9.9 months). Seven patients in each arm had an objective response.
Glioblastoma: AVF3708g: The efficacy and safety of Bevacizumab as treatment for patients with glioblastoma was studied in an open-label, multicentre, randomized, non-comparative study (study AVF3708g).
Glioblastoma patients in first or second relapse after prior radiotherapy (completed at least 8 weeks prior to receiving Bevacizumab) and temozolomide, were randomized (1:1) to receive Bevacizumab (10 mg/kg IV infusion every 2 weeks) or Bevacizumab plus irinotecan (125 mg/m2 IV or 340 mg/m2 IV for patients on enzyme-inducing antiepileptic drugs every 2 weeks) until disease progression or until unacceptable toxicity. The primary endpoints of the study were 6-month progression-free survival (PFS) and objective response rate (ORR) as assessed by an independent review facility (IRF). Other outcome measures were duration of PFS, duration of response and overall survival.
Results of the study are summarized in Table 10. (See Table 10.)
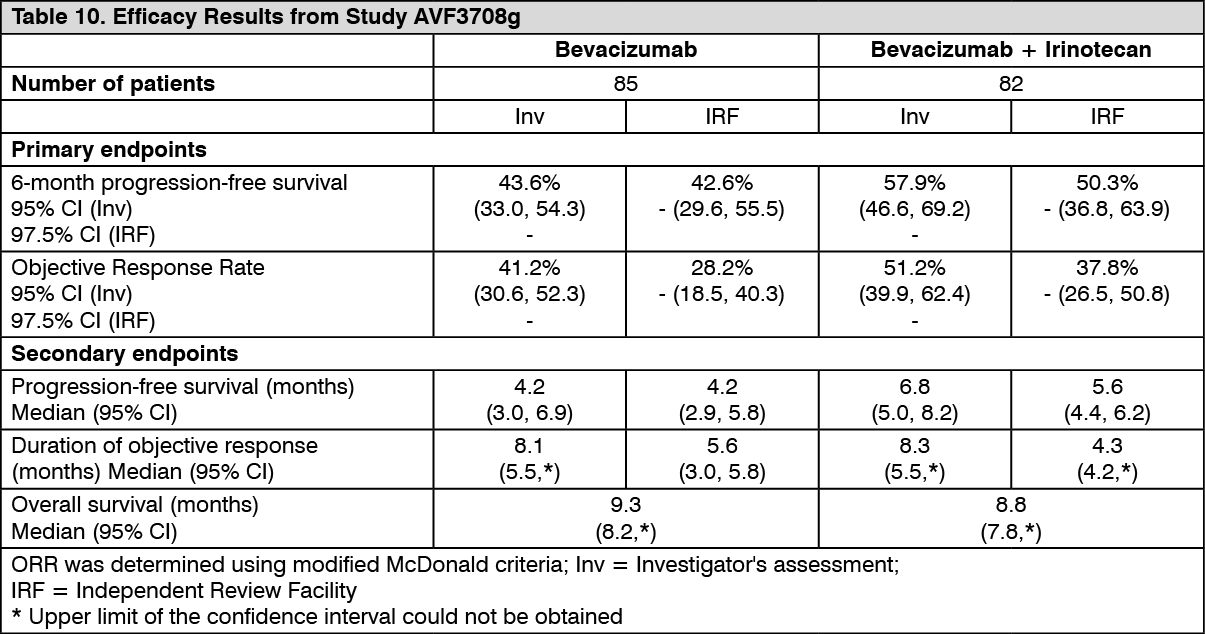 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn study AVF3708g, six-month PFS based on IRF assessments was significantly higher (p<0.0001) compared with historical controls for both treatment arms: 42.6% in the Bevacizumab arm and 50.3% in the Bevacizumab plus irinotecan arm (investigator assessment: 43.6% in the Bevacizumab arm and 57.9% in the Bevacizumab plus irinotecan arm). Objective response rates were also significantly higher (p<0.0001) compared with historical controls for both treatment arms: 28.2% in the Bevacizumab arm and 37.8% in the Bevacizumab plus irinotecan arm (investigator assessment: 41.2% in the Bevacizumab arm and 51.2% in the Bevacizumab plus irinotecan arm).
The majority of patients who were receiving steroids at baseline, including responders and non-responders, were able to reduce their steroid utilization over time while receiving bevacizumab treatment. The majority of patients experiencing an objective response or prolonged PFS (at week 24) were able to maintain or improve their neurocognitive functions while on study treatment compared to baseline. The majority of patients that remained in the study and were progression free at 24 weeks, had a Karnofsky performance status (KPS) that remained stable.
Epithelial Ovarian, Fallopian Tube and Primary Peritoneal Cancer: Front-line Ovarian Cancer: The safety and efficacy of Bevacizumab in the front-line treatment of patients with epithelial ovarian, fallopian tube or primary peritoneal cancer were studied in two phase III trials (GOG-0218 and BO17707) that compared the effect of the addition of Bevacizumab to carboplatin and paclitaxel compared to the chemotherapy regimen alone.
GOG-0218The GOG-0218 study was a Phase III multicenter, randomized, double-blind, placebo-controlled, three-arm study evaluating the effect of adding Bevacizumab to an approved chemotherapy regimen (carboplatin and paclitaxel) in patients with optimally or suboptimally debulked Stage III or Stage IV epithelial ovarian, fallopian tube or primary peritoneal cancer.
A total of 1873 patients were randomized in equal proportions to the following three arms: CPP arm: Placebo in combination with carboplatin (AUC 6) and paclitaxel (175 mg/m2) for 6 cycles followed by placebo alone, for a total of up to 15 months of therapy; CPB15 arm: Five cycles of Bevacizumab (15 mg/kg q3w) in combination with carboplatin (AUC 6) and paclitaxel (175 mg/m2) for 6 cycles (Bevacizumab commenced at cycle 2 of chemotherapy) followed by placebo alone, for a total of up to 15 months of therapy; CPB15+ arm: Five cycles of Bevacizumab (15 mg/kg q3w) in combination with carboplatin (AUC 6) and paclitaxel (175 mg/m2) for 6 cycles (Bevacizumab commenced at cycle 2 of chemotherapy) followed by continued use of Bevacizumab (15 mg/kg q3w) as single agent for a total of up to 15 months of therapy.
The primary endpoint was Progression Free Survival (PFS) based on investigator's assessment of radiological scans. In addition, an independent review of the primary endpoint was also conducted. The results of this study are summarized in Table 11. (See Table 11.)
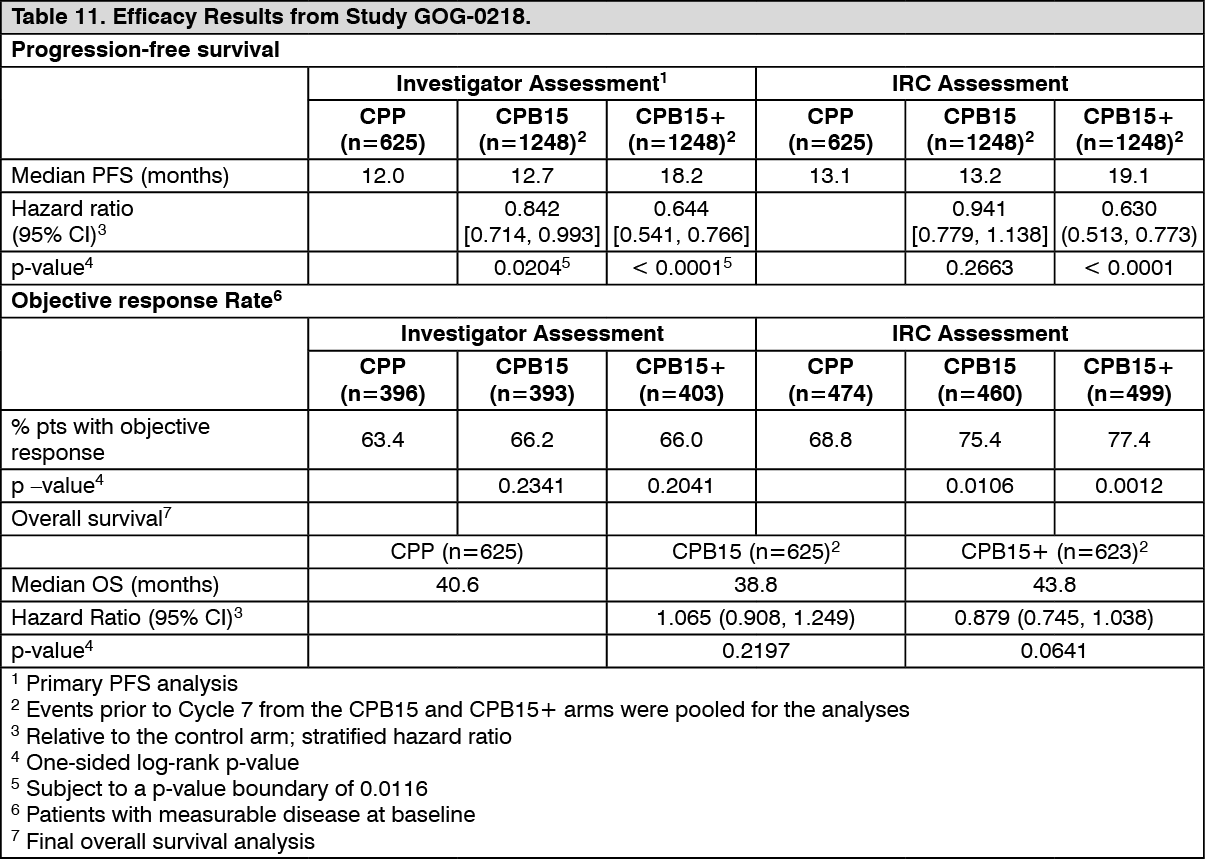 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe trial met its primary objective of PFS improvement. Compared to patients treated with chemotherapy (carboplatin and paclitaxel) alone, patients who received front-line bevacizumab at a dose of 15 mg/kg q3w in combination with chemotherapy and continued to receive bevacizumab alone, had a clinically meaningful and statistically significant improvement in PFS.
Although there was an improvement in PFS for patients who received front-line bevacizumab in combination with chemotherapy and did not continue to receive bevacizumab alone, the improvement was neither clinically meaningful nor statistically significant compared to patients who received chemotherapy alone.
BO17707 (ICON7): BO17707 was a Phase III, two arm, multicenter, randomized, controlled, open-label study comparing the effects of adding Bevacizumab to carboplatin plus paclitaxel in patients with FIGO Stage I or IIA (Grade 3 or clear cell histology only), or FIGO Stage IIB - IV (all grades and all histological types) epithelial ovarian, fallopian tube or primary peritoneal cancer following surgery, and in whom no further surgery was planned before progression.
A total of 1528 patients were randomized in equal proportions to the following two arms: CP arm: Carboplatin (AUC 6) and paclitaxel (175 mg/m2) for 6 cycles; CPB7.5+ arm: Carboplatin (AUC 6) and paclitaxel (175 mg/m2) for 6 cycles plus Bevacizumab (7.5 mg/kg q3w) for up to 18 cycles.
The primary endpoint was Progression Free Survival (PFS) as assessed by the investigator.
The results of this study are summarized in Table 12. (See Table 12.)
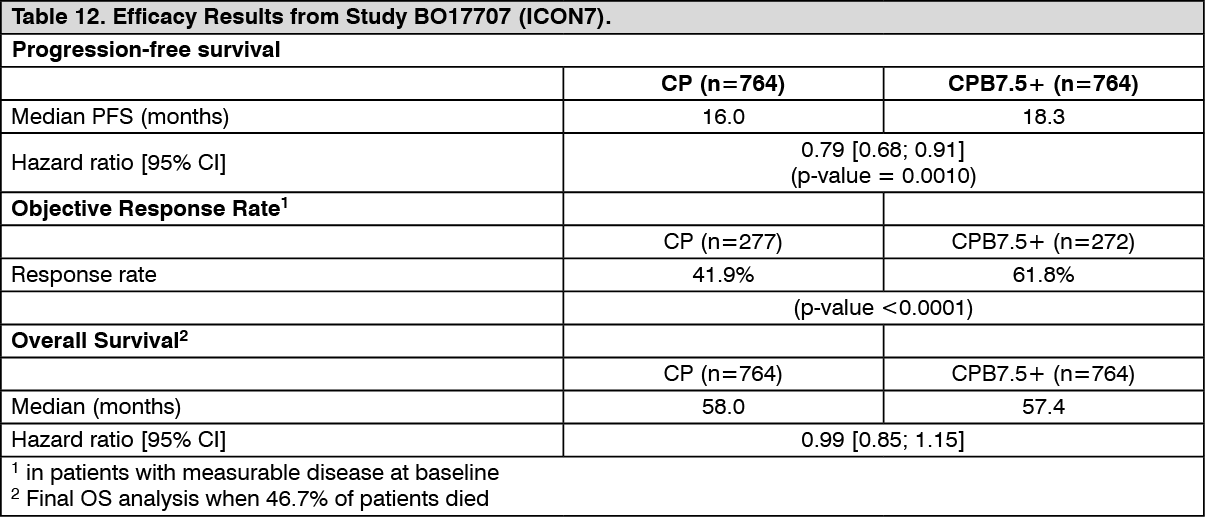 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe trial met its primary objective of PFS improvement. Compared to patients treated with chemotherapy (carboplatin and paclitaxel) alone, patients who received bevacizumab at a dose of 7.5 mg/kg q3w in combination with chemotherapy and continued to receive bevacizumab for up to 18 cycles had a statistically significant improvement in PFS.
Recurrent Ovarian Cancer: GOG-0213: GOG-0213 was a phase III randomized controlled trial studying the safety and efficacy of Bevacizumab in the treatment of patients with platinum-sensitive, recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer, who have not received prior chemotherapy in the recurrent setting. There was no exclusion criterion for prior anti-angiogenic therapy.
The study evaluated the effect of adding Bevacizumab to carboplatin + paclitaxel and continuing Bevacizumab as a single agent compared to carboplatin + paclitaxel alone.
A total of 673 patients were randomized in equal proportions to the following two treatment arms. CP arm: Carboplatin (AUC5) and paclitaxel (175 mg/m2 IV over 3 hours) every 3 weeks for 6 and up to 8 cycles; CPB arm: Carboplatin (AUC5) and paclitaxel (175 mg/m2 IV over 3 hours) and concurrent Bevacizumab (15 mg/kg) every 3 weeks for 6 and up to 8 cycles followed by Bevacizumab (15 mg/kg every 3 weeks) alone until disease progression or unacceptable toxicity.
The primary efficacy endpoint was overall survival (OS). The main secondary efficacy endpoint was progression-free survival (PFS). Objective response rates (ORR) were also examined. Results are presented in Table 13. (See Table 13.)
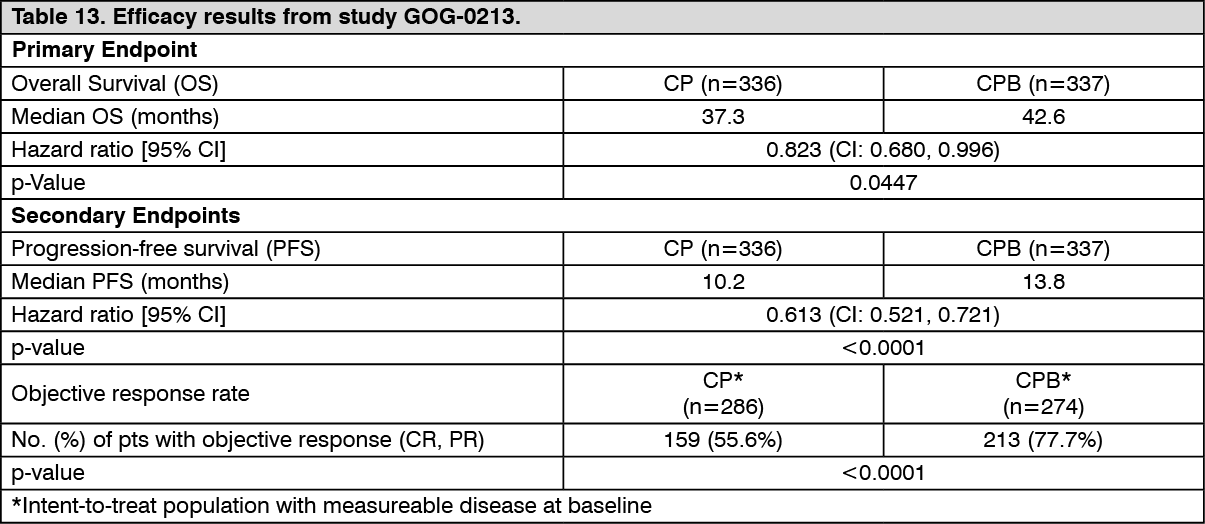 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment with Bevacizumab at 15 mg/kg every 3 weeks in combination with chemotherapy (carboplatin and paclitaxel) for 6 and up to 8 cycles then followed by Bevacizumab as a single agent resulted in a clinically meaningful and statistically significant improvement in OS compared to treatment with carboplatin and paclitaxel alone.
AVF4095g: The safety and efficacy of Bevacizumab in the treatment of patients with platinum-sensitive, recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer, who have not received prior chemotherapy in the recurrent setting or prior bevacizumab treatment, was studied in a phase III trial randomized, double-blind, placebo-controlled trial (AVF4095g).
The study compared the effect of adding Bevacizumab to carboplatin and gemcitabine chemotherapy and continuing Bevacizumab as a single agent to progression to carboplatin and gemcitabine alone.
A total of 484 patients with measurable disease were randomized in equal portions to either: Carboplatin (AUC4, Day 1) and gemcitabine (1000 mg/m2 on Days 1 and 8) and concurrent placebo every 3 weeks for 6 and up to 10 cycles followed by placebo alone until disease progression or unacceptable toxicity; Carboplatin (AUC4, Day 1) and gemcitabine (1000 mg/m2 on Days 1 and 8) and concurrent Bevacizumab (15 mg/kg Day 1) every 3 weeks for 6 and up to 10 cycles followed by Bevacizumab (15 mg/kg every 3 weeks) alone until disease progression or unacceptable toxicity.
The primary endpoint was progression-free survival based on investigator assessment using RECIST criteria. Additional endpoints included objective response, duration of response, safety and overall survival. An independent review of the primary endpoint was also conducted.
The results of this study are summarized in Table 14. (See Table 14.)
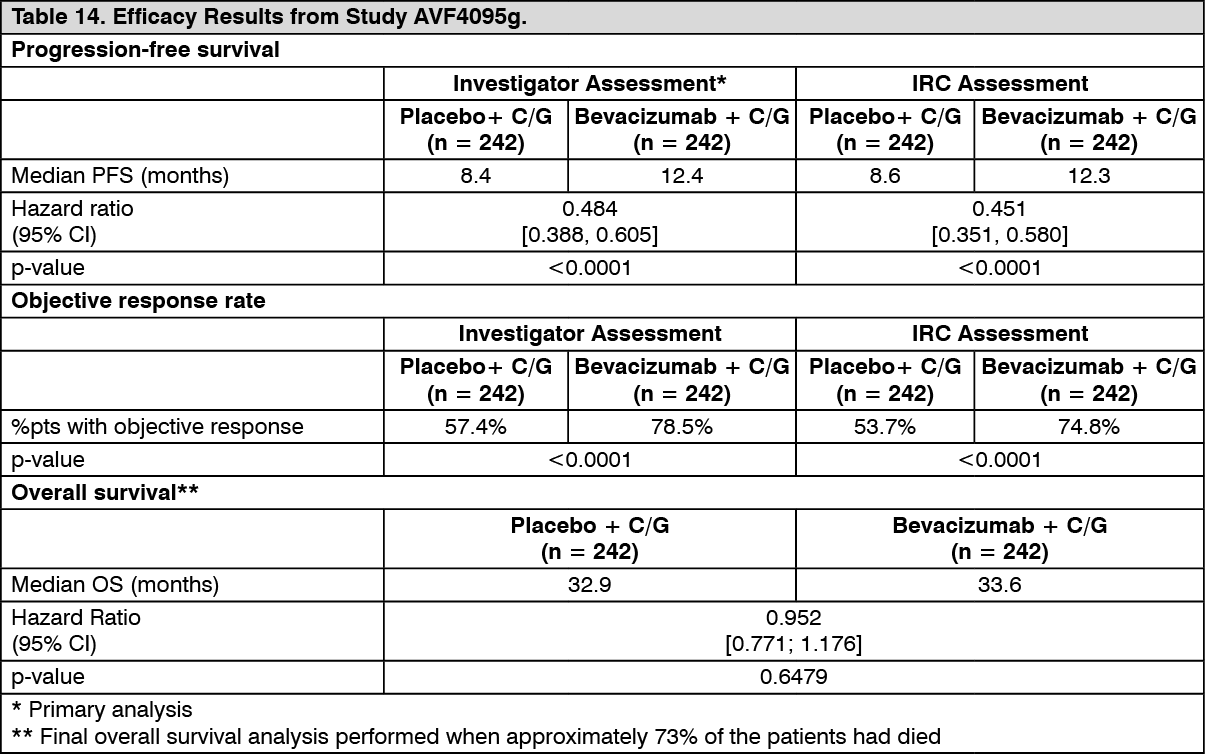 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageMO22224 (AURELIA): Study MO22224 evaluated the efficacy and safety of bevacizumab in combination with chemotherapy for platinum-resistant recurrent ovarian cancer. This study was designed as an open-label, randomized, two-arm Phase III evaluation of bevacizumab plus chemotherapy (CT+BV) versus chemotherapy alone (CT).
A total of 361 patients were enrolled into this study and administered either chemotherapy (paclitaxel, topotecan, or PLD) alone or in combination with bevacizumab: CT Arm (chemotherapy alone): Paclitaxel 80 mg/m2 as a 1-hour IV infusion on Days 1, 8, 15, and 22 every 4 weeks; Topotecan 4 mg/m2 as a 30-minute IV infusion on Days 1, 8, and 15 every 4 weeks. Alternatively, a 1.25 mg/m2 dose could be administered over 30 minutes on Days 1-5 every 3 weeks; PLD 40 mg/m2 as a 1 mg/min IV infusion on Day 1 only every 4 weeks.
After Cycle 1, the drug could be delivered as a 1-hour infusion. CT+BV Arm (chemotherapy plus bevacizumab): The chosen chemotherapy was combined with bevacizumab 10 mg/kg IV every 2 weeks (or bevacizumab 15 mg/kg every 3 weeks if used in combination with topotecan 1.25 mg/m2 on Days 1-5 on a every 3 weeks schedule).
Eligible patients had ovarian cancer that progressed within 6 months of previous platinum therapy. If a patient had been previously included in a blinded trial with an antiangiogenic agent, the patient was enrolled in the same stratum as those patients who were known to have previously received an anti-angiogenic agent.
The primary endpoint was progression-free survival, with secondary endpoints including objective response rate and overall survival. Results are presented in Table 15. (See Table 15.)
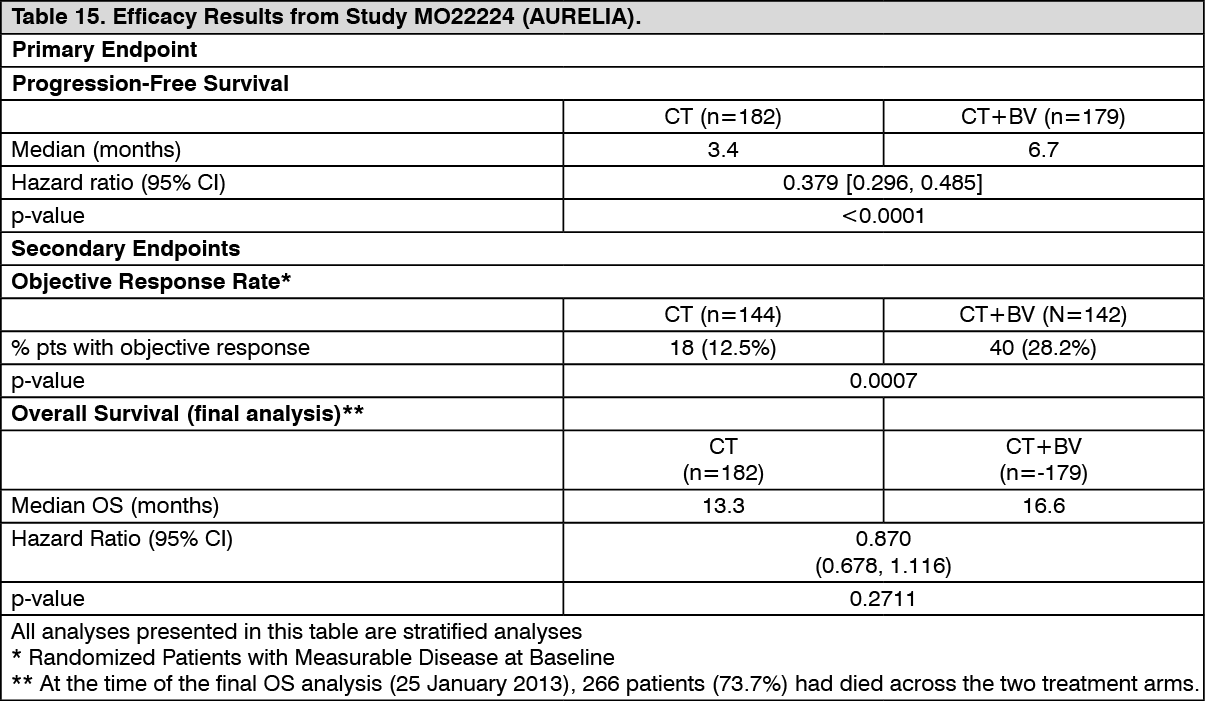 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageCervical Cancer: GOG-0240: The efficacy and safety of bevacizumab in combination with chemotherapy (paclitaxel and cisplatin or paclitaxel and topotecan) as a treatment for patients with persistent, recurrent, or metastatic carcinoma of the cervix was evaluated in study GOG-0240, a randomized, four-arm, multi-centre phase III trial.
A total of 452 patients were randomized to receive either: Paclitaxel 135 mg/m2 IV over 24 hours on Day 1 and cisplatin 50 mg/m2 IV on Day 2, every 3 weeks (q3w); or paclitaxel 175 mg/m2 IV over 3 hours on Day 1 and cisplatin 50 mg/m2 IV on Day 2 (q3w); or paclitaxel 175 mg/m2 IV over 3 hours on Day 1 and cisplatin 50 mg/m2 IV on Day 1 (q3w); Paclitaxel 135 mg/m2 IV over 24 hours on Day 1 and cisplatin 50 mg/m2 IV on Day 2 plus bevacizumab 15 mg/kg IV on Day 2 (q3w); or paclitaxel 175 mg/m2 IV over 3 hours on Day 1 and cisplatin 50 mg/m2 IV on Day 2 plus bevacizumab 15 mg/kg IV on Day 2 (q3w); or paclitaxel 175 mg/m2 IV over 3 hours on Day 1 and cisplatin 50 mg/m2 IV on Day 1 and bevacizumab 15 mg/kg IV on Day 1 (q3w); Paclitaxel 175 mg/m2 over 3 hours on Day 1 and topotecan 0.75 mg/m2 over 30 minutes on days 1-3 (q3w); Paclitaxel 175 mg/m2 over 3 hours on Day 1 and topotecan 0.75 mg/m2 over 30 minutes on Days 1-3 plus bevacizumab 15 mg/kg IV on Day 1 (q3w).
Eligible patients had persistent, recurrent, or metastatic squamous cell carcinoma, adenosquamous carcinoma, or adenocarcinoma of the cervix which was not amenable to curative treatment with surgery and/or radiation therapy.
The primary efficacy endpoint was overall survival (OS). Secondary efficacy endpoints included progression-free survival (PFS) and objective response rate (ORR). Results are presented in Table 16. (See Table 16.)
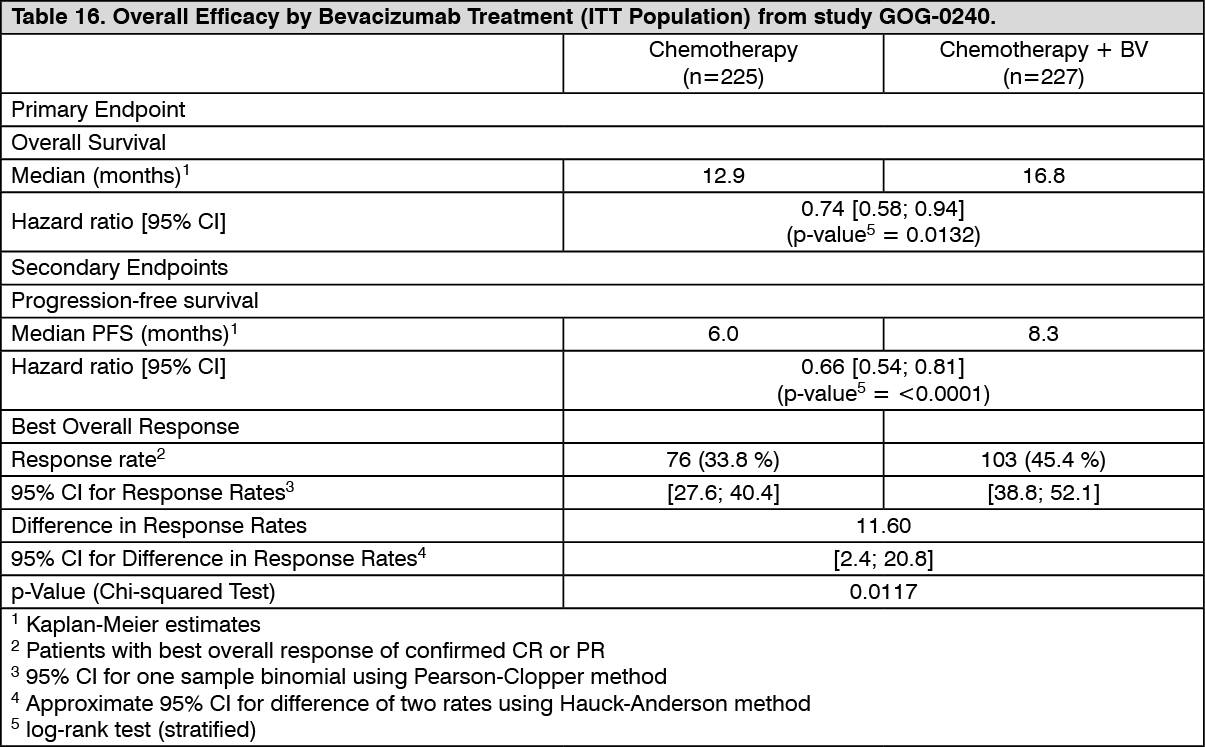 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageImmunogenicity: No robust assessment of anti-drug antibodies has been done in Bevacizumab clinical trials.
Pharmacokinetics: The pharmacokinetics of bevacizumab were characterised in patients with various types of solid tumours. The doses tested were 0.1-10 mg/kg weekly in phase I; 3-20 mg/kg every two weeks (q2w) or every three weeks (q3w) in phase II; 5 mg/kg (q2w) or 15 mg/kg q3w in phase III. In all clinical trials, bevacizumab was administered as an IV infusion.
As observed with other antibodies, the pharmacokinetics of bevacizumab are well described by a two-compartment model. Overall, in all clinical trials, bevacizumab disposition was characterized by a low clearance, a limited volume of the central compartment (Vc), and a long elimination half-life. This enables target therapeutic bevacizumab serum levels to be maintained with a range of administration schedules (such as one administration every 2 or 3 weeks).
In the population pharmacokinetics analysis there was no significant difference in the pharmacokinetics of bevacizumab in relation to age (no correlation between bevacizumab clearance and patient age [the median age was 59 years with 5th and 95th percentiles of 37 and 76 years]).
Low albumin and high tumour burden are generally indicative of disease severity.
Bevacizumab clearance was approximately 30% faster in patients with low levels of serum albumin and 7% faster in subjects with higher tumour burden when compared with a typical patient with median values of albumin and tumour burden.
Study MYL 1402O 1002: A double-blind, single-dose, three-treatment, parallel group study comparing PK of KRABEVA to US-Avastin and EU-Avastin (bevacizumab) in a total of 111 healthy, adult male volunteers (37 subjects per treatment arm). After randomization, subjects received a single 1 mg/kg dose of study treatment administered by iv infusion (25 mL over approximately 90 minutes). Pharmacokinetics following the single administration derived from the serum concentration-time curves were analysed using analysis of variance.
The statistical analysis for the primary PK parameter, AUC0-inf, as well as for the secondary PK parameters, Cmax and AUC0-t, reveals that the 90% CIs fall within 80% to 125% for the test to reference ratio for the natural log transformed parameters, LNAUC0-inf, LNAUC0-t and LNCmax, for bevacizumab for all 3 pairwise comparisons.
This study demonstrated that KRABEVA is bioequivalent to US-Avastin and EU-Avastin, and that EU-Avastin is bioequivalent to US-Avastin.
BM100-CC-03-I-01: The mean plasma concentration profiles of KRABEVA and Avastin were similar over time. The point estimates of the ratio of least square means of Bmab-100 and Avastin for the log-transformed primary PK parameters C and AUC (AUC) were close to 1; showing PK similarity. The 90% CIs of the ratios were within the predefined max 0-t 0-504 bioequivalence range of 80.00% to 125.00% (Cmax: 85.86% to 100.54%; and AUC0-504: 87.56% to 104.79%). This study demonstrated bioequivalence between KRABEVA and Avastin.
Absorption: No text.
Distribution: The typical value for central volume (Vc) was 2.73 L and 3.28 L for female and male subjects respectively, which is in the range that has been described for IgGs and other monoclonal antibodies. The typical value for peripheral volume (Vp) was 1.69 L and 2.35 L for female and male patients respectively, when bevacizumab is co-administered with antineoplastic agents. After correcting for body weight, male subjects had a larger Vc (+20%) than females.
Metabolism: Assessment of bevacizumab metabolism in rabbits following a single i.v. dose of 125 I bevacizumab indicated that its metabolic profile was similar to that expected for a native IgG molecule which does not bind VEGF. The metabolism and elimination of bevacizumab is similar to endogenous IgG i.e. primarily via proteolytic catabolism throughout the body, including endothelial cells, and does not rely primarily on elimination through the kidneys and liver. Binding of the IgG to the FcRn receptor result in protection from cellular metabolism and the long terminal half-life.
Elimination: The pharmacokinetics of bevacizumab are linear at doses ranging from 1.5 to 10 mg/kg/wk.
The value for clearance is, on average, equal to 0.188 and 0.220 L/day for female and male patients, respectively. After correcting for body weight, male patients had a higher bevacizumab clearance (+17%) than females. According to the two-compartmental model, the elimination half-life is 18 days for a typical female patient and 20 days for a typical male patient.
Pharmacokinetics in Special Populations: The population pharmacokinetics of bevacizumab were analysed to evaluate the effects of demographic characteristics. In adults, the results showed no significant difference in the pharmacokinetics of bevacizumab in relation to age.
Pediatric population: The pharmacokinetics of bevacizumab were evaluated in 152 patients (7 months to 21 years; 5.9 to 125 kg) across 4 clinical studies using a population pharmacokinetic model. The pharmacokinetic results show that the clearance and the volume of distribution of bevacizumab were comparable between pediatric and adult patients when normalized by body weight. Age was not associated with the pharmacokinetics of bevacizumab when body weight was taken into account.
Renal impairment: No studies have been conducted to investigate the pharmacokinetics of bevacizumab in renally impaired patients since the kidneys are not a major organ for bevacizumab metabolism or excretion.
Hepatic impairment: No studies have been conducted to investigate the pharmacokinetics of bevacizumab in patients with hepatic impairment since the liver is not a major organ for bevacizumab metabolism or excretion.
Toxicology: Nonclinical Safety: Carcinogenicity: Studies have not been performed to evaluate the carcinogenic potential of Bevacizumab.
Genotoxicity: Studies have not been performed to evaluate the mutagenic potential of Bevacizumab.
Impairment of Fertility: No specific studies in animals have been performed to evaluate the effect of Bevacizumab on fertility. No adverse effect on male reproductive organs was observed in repeat dose toxicity studies in cynomolgus monkeys.
Inhibition of ovarian function was characterised by decreases in ovarian and/or uterine weight and the number of corpora lutea, a reduction in endometrial proliferation and an inhibition of follicular maturation in cynomolgus monkeys treated with Bevacizumab for 13 or 26 weeks.
The doses associated with this effect were ≥4 times the human therapeutic dose or ≥2-fold above the expected human exposure based on average serum concentrations in female monkeys. In rabbits, administration of 50 mg/kg of bevacizumab resulted in a significant decrease in ovarian weight and number of corpora lutea. The results in both monkeys and rabbits were reversible upon cessation of treatment. The inhibition of angiogenesis following administration of bevacizumab is likely to result in an adverse effect on female fertility.
Reproductive Toxicity: Bevacizumab has been shown to be embryotoxic and teratogenic when administered to rabbits. Observed effects included decreases in maternal and foetal body weights, an increased number of foetal resorptions and an increased incidence of specific gross and skeletal foetal alterations. Adverse foetal outcomes were observed at all tested doses of 10-100 mg/kg.
Information on foetal malformations observed in the postmarketing setting are provided in Use in Pregnancy & Lactation and Postmarketing Experience under Adverse Reactions.
Other: Physeal development: In studies of up to 26 weeks duration in cynomolgus monkeys, Bevacizumab was associated with physeal dysplasia. Physeal dysplasia was characterised primarily by thickened growth plate cartilage, subchondral bony plate formation and inhibition of vascular invasion of the growth plate. This effect occurred at doses ≥0.8 times the human therapeutic dose and exposure levels slightly below the expected human clinical exposure, based on average serum concentrations. It should be noted, however, that physeal dysplasia occurred only in actively growing animals with open growth plates.
Wound healing: In rabbits, the effects of bevacizumab on circular wound healing were studied. Wound reepithelialisation was delayed in rabbits following five doses of bevacizumab, ranging from 2-50 mg/kg, over a 2-week period. A trend toward a dose-dependent relationship was observed. The magnitude of effect on wound healing was similar to that observed with corticosteroid administration. Upon treatment cessation with either 2 or 10 mg/kg bevacizumab, the wounds closed completely. The lower dose of 2 mg/kg was approximately equivalent to the proposed clinical dose. A more sensitive linear wound healing model was also studied in rabbits. Three doses of bevacizumab ranging from 0.5-2 mg/kg dose-dependently and significantly decreased the tensile strength of the wounds, consistently with delayed wound healing. The low dose of 0.5 mg/kg was 5-fold below the proposed clinical dose.
As effects on wound healing were observed in rabbits at doses below the proposed clinical dose, the capacity for bevacizumab to adversely impact wound healing in human should be considered.
In cynomolgus monkeys, the effects of bevacizumab on the healing of a linear incision were highly variable and no dose-response relationship was evident.
Renal function: In normal cynomolgus monkeys, bevacizumab had no measurable effect on renal function treated once or twice weekly for up to 26 weeks, and did not accumulate in the kidney of rabbits following two doses up to 100 mg/kg (approximately 80-fold the proposed clinical dose).
Investigative toxicity studies in rabbits, using the models of renal dysfunction, showed that bevacizumab did not exacerbate renal glomerular injury induced by bovine serum albumin or renal tubular damage induced by cisplatin.
Albumin: In male cynomolgus monkeys, Bevacizumab administered at doses of 10 mg/kg twice weekly or 50 mg/kg once weekly for 26 weeks was associated with a statistically significant decrease in albumin and albumin to globulin ratio and increase in globulin. These effects were reversible upon cessation of exposure. As the parameters remained within the normal reference range of values for these endpoints, these changes were not considered as clinically significant.
Hypertension: At doses up to 50 mg/kg twice weekly in cynomolgus monkeys, Bevacizumab showed no effects on blood pressure.
Haemostasis: Nonclinical toxicology studies of up to 26 weeks duration in cynomolgus monkeys did not find changes in hematology or coagulation parameters including platelet counts, prothrombin and activated partial thromboplastin time. A model of hemostasis in rabbits, used to investigate the effect of bevacizumab on thrombus formation, did not show alteration in the rate of clot formation or any other hematological parameters compared to treatment with Bevacizumab vehicle.