Product Specific Warnings and Precautions: Increased Mortality in Elderly Patients with Dementia-related Psychosis: Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death compared to placebo. Analyses of 17 placebo-controlled trials (modal duration of 10 weeks), in patients taking atypical antipsychotic drugs (including risperidone, aripiprazole, olanzapine, and quetiapine), revealed a risk of death in drug-treated patients of between 1.6 to 1.7 times the risk of death in placebo-treated patients. Over the course of a typical 10-week controlled trial, the rate of death in drug treated patients was about 4.5%, compared to a rate of about 2.6% in the placebo group.
Although the causes of death varied, most of the deaths appeared to be either cardiovascular (e.g., heart failure, sudden death) or infectious (e.g., pneumonia) in nature. Brexpiprazole is not approved for the treatment of dementia-related psychosis.
Cerebrovascular Adverse Reactions Including Stroke in Elderly Patients with Dementia-related Psychosis: In placebo-controlled trials in elderly subjects with dementia, patients randomized to risperidone, aripiprazole, and olanzapine had a higher incidence of stroke and transient ischemic attack, including fatal stroke. REXULTI is not approved for the treatment of patients with dementia-related psychosis.
Suicidal Risk: The possibility of a suicide attempt is inherent in psychotic illnesses and major depressive disorder (MDD). Close supervision and appropriate clinical management of high-risk patients should accompany drug therapy.
Suicidal Thoughts and Behaviours in Children, Adolescents and Young Adults: In pooled analyses of placebo-controlled trials of antidepressant drugs (SSRIs and other antidepressant classes) that included approximately 77,000 adult patients, and over 4,400 paediatric patients, the incidence of suicidal thoughts and behaviours in patients age 24 years and younger was greater in antidepressant-treated patients than in placebo-treated patients. The drug-placebo differences in the number of cases of suicidal thoughts and behaviours per 1000 patients treated are provided in Table 5. No suicides occurred in any of the paediatric studies. There were suicides in the adult studies, but the number was not sufficient to reach any conclusion about antidepressant drug effect on suicide. (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
It is unknown whether the risk of suicidal thoughts and behaviours in children, adolescents, and young adults extends to longer-term use, i.e., beyond four months. However, there is substantial evidence from placebo-controlled maintenance studies in adults with MDD that antidepressants delay the recurrence of depression.
Monitor all antidepressant-treated patients for clinical worsening and emergence of suicidal thoughts and behaviours, especially during the initial few months of drug therapy and at times of dosage changes. Counsel family members or caregivers of patients to monitor for changes in behaviour and to alert the healthcare provider. Consider changing the therapeutic regimen, including possibly discontinuing REXULTI, in patients whose depression is persistently worse, or who are experiencing emergent suicidal thoughts or behaviours.
Neuroleptic Malignant Syndrome (NMS): A potentially fatal symptom complex, referred to as Neuroleptic Malignant Syndrome (NMS), has been reported in association with administration of antipsychotic drugs including REXULTI. Clinical manifestations of NMS are hyperpyrexia, muscle rigidity, altered mental status and evidence of autonomic instability (irregular pulse or blood pressure, tachycardia, diaphoresis and cardiac dysrhythmia). Additional signs may include elevated creatine phosphokinase, myoglobinuria (rhabdomyolysis), and acute renal failure. If a patient develops signs and symptoms indicative of NMS, or presents with unexplained high fever without additional clinical manifestations of NMS, all antipsychotic drugs including REXULTI must be discontinued. If a patient requires antipsychotic drug treatment after recovery from NMS, the potential reintroduction of drug therapy should be carefully considered.
Tardive Dyskinesia: Tardive dyskinesia, a syndrome consisting of potentially irreversible, involuntary, dyskinetic movements may develop in patients treated with antipsychotic drugs. The risk appears to be highest among the elderly, especially elderly women, but it is not possible to predict which patients are likely to develop the syndrome. Whether antipsychotic drugs differ in their potential to cause tardive dyskinesia is unknown.
The risk of tardive dyskinesia and the likelihood that it will become irreversible increase with the duration of treatment and the cumulative dose. The syndrome can develop after a relatively brief treatment period, even at low doses. It may also occur after discontinuation of treatment.
There is no known treatment for established cases of tardive dyskinesia, although the syndrome may remit, partially or completely, if antipsychotic treatment is discontinued. Antipsychotic treatment itself, however, may suppress (or partially suppress) the signs and symptoms of the syndrome, possibly masking the underlying process. The effect that symptomatic suppression has upon the long-term course of the syndrome is unknown.
Given these considerations, REXULTI should be prescribed in a manner most likely to reduce the risk of tardive dyskinesia. Chronic antipsychotic treatment should generally be reserved for patients: who suffer from a chronic illness that is known to respond to antipsychotic drugs; and for whom alternative, effective, but potentially less harmful treatments are not available or appropriate. In patients who do require chronic treatment, use the lowest dose and the shortest duration of treatment needed to produce a satisfactory clinical response. Periodically reassess the need for continued treatment.
If signs and symptoms of tardive dyskinesia appear in a patient on REXULTI, drug discontinuation should be considered. However, some patients may require treatment with REXULTI despite the presence of the syndrome.
Metabolic Changes: Atypical antipsychotic drugs, including REXULTI, have caused metabolic changes, including hyperglycemia, diabetes mellitus, dyslipidemia, and body weight gain. Although all of the drugs in the class to date have been shown to produce some metabolic changes, each drug has its own specific risk profile.
Hyperglycemia and Diabetes Mellitus: Hyperglycemia, in some cases extreme and associated with ketoacidosis or hyperosmolar coma or death, has been reported in patients treated with atypical antipsychotics. Assessment of the relationship between atypical antipsychotics use and glucose abnormalities is complicated by the possibility of an increased background risk of diabetes mellitus in patients with schizophrenia and the increasing incidence of diabetes mellitus in the general population. Given these confounders, the relationship between atypical antipsychotic use and hyperglycaemia-related adverse events is not completely understood. However, epidemiological studies suggest an increased risk of treatment-emergent hyperglycaemia-related adverse events in patients treated with the atypical antipsychotics. Precise risk estimates for hyperglycaemia-related adverse events in patients treated with atypical antipsychotics are not available.
Patients with an established diagnosis of diabetes mellitus who are started on atypical antipsychotics should be monitored regularly for worsening of glucose control. Patients with risk factors for diabetes mellitus (e.g. obesity, family history of diabetes) who are starting treatment with atypical antipsychotics should undergo fasting blood glucose testing in the beginning of treatment and periodically during treatment. Any patient treated with atypical antipsychotics should be monitored for symptoms of hyperglycaemia including polydipsia, polyuria, polyphagia, and weakness. Patients who develop symptoms of hyperglycaemia during treatment with atypical antipsychotics should undergo fasting blood-glucose testing. In some cases, hyperglycaemia has resolved when the atypical antipsychotic was discontinued; however, some patients required continuation of anti-diabetic treatment despite discontinuation of the suspect drug.
There have been reports of hyperglycemia in patients treated with REXULTI. (see Adverse Reactions).
Dyslipidemia: Atypical antipsychotics cause adverse alterations in lipids. Before or soon after initiation of antipsychotic medication, obtain a fasting lipid profile at baseline and monitor periodically during treatment (see Adverse Reactions).
Weight Gain: Weight gain has been observed in patients treated with atypical antipsychotics, including REXULTI. Monitor weight at baseline and frequently thereafter.
For more details related to metabolic changes and weight gain see Adverse Reactions.
Prolactin: REXULTI can elevate prolactin levels. Elevations associated with REXULTI treatment are generally mild and may decline during administration, however, in some infrequent cases the effect may persist during administration.
Genitourinary: Although no cases of priapism were reported in clinical trials with REXULTI, rare cases of priapism have been reported with antipsychotic use. With other psychotropic drugs, this adverse reaction did not appear to be dose-dependent and did not correlate with the duration of treatment.
Venous thromboembolism: Venous thromboembolism (VTE), including fatal pulmonary embolism, has been reported with antipsychotic drugs including REXULTI, in case reports and/or observational studies. When prescribing REXULTI all potential risk factors for VTE should be identified and preventative measures undertaken.
Immune: Hypersensitivity: Spontaneous post-market reports of serious hypersensitivity reactions, such as anaphylaxis, angioedema and facial swelling, rash and urticaria, have been reported with REXULTI.
Cardiovascular disorders: REXULTI has not been evaluated in patients with a history of myocardial infarction/ischaemic heart disease or clinically significant cardiovascular disease since such patients were excluded from clinical trials.
REXULTI should be used with caution in patients with known cardiovascular disease (history of myocardial infarction or ischaemic heart disease, heart failure, or conduction abnormalities), cerebrovascular disease, conditions which would predispose patients to hypotension (dehydration, hypovolemia, and treatment with antihypertensive medicinal products) or hypertension (including accelerated or malignant).
Leukopenia, Neutropenia and Agranulocytosis: Leukopenia and neutropenia have been reported during treatment with antipsychotic agents. Agranulocytosis (including fatal cases) has been reported with other agents in this class.
Possible risk factors for leukopenia and neutropenia include pre-existing low white blood cell count (WBC) or absolute neutrophil count (ANC) and history of drug-induced leukopenia or neutropenia. In patients with a pre-existing low WBC or ANC or a history of drug-induced leukopenia or neutropenia, perform a complete blood count (CBC) frequently during the first few months of therapy. In such patients, consider discontinuation of REXULTI at the first sign of a clinically significant decline in WBC in the absence of other causative factors.
Monitor patients with clinically significant neutropenia for fever or other symptoms or signs of infection and treat promptly if such symptoms or signs occur. Discontinue REXULTI in patients with absolute neutrophil count <1000/mm
3 and follow their WBC until recovery.
Orthostatic Hypotension and Syncope: Atypical antipsychotics cause orthostatic hypotension and syncope. Generally, the risk is greatest during initial dose titration and when increasing the dose.
In two short-term, placebo-controlled clinical studies of REXULTI+ADT in patients with MDD Studies 1 (Study 228) and 2 (Study 227), the incidence of orthostatic hypotension-related adverse reactions in REXULTI+ADT-treated patients compared to placebo+ADT patients included: dizziness (2% vs. 2%) and orthostatic hypotension (0.1% vs. 0%). In two short-term, placebo-controlled clinical studies of REXULTI in patients with schizophrenia Studies 6 (Study 231) and 7 (Study 230) the incidence of orthostatic hypotension-related adverse reactions in REXULTI-treated patients compared to placebo patients included: dizziness (2% versus 2%), orthostatic hypotension (0.4% versus 0.2%), and syncope (0.1% versus 0%).
Orthostatic vital signs should be monitored in patients who are vulnerable to hypotension, (e.g., elderly patients, patients with dehydration, hypovolemia, concomitant treatment with antihypertensive medication), patients with known cardiovascular disease (history of myocardial infarction, ischemic heart disease, heart failure, or conduction abnormalities), and patients with cerebrovascular disease. REXULTI has not been evaluated in patients with a recent history of myocardial infarction or unstable cardiovascular disease. Such patients were excluded from pre-marketing clinical trials.
QT Interval: QT prolongation can develop in patients treated with antipsychotics. In clinical trials, only a few, nonserious, QT prolongations have been reported with REXULTI. Caution should be exercised when REXULTI is prescribed in patients with known cardiovascular disease, family history of QT prolongation, electrolyte imbalance or in concomitant use with other medicinal products thought to prolong the QT interval.
Falls: Antipsychotics, including REXULTI, may cause somnolence, postural hypotension, motor and sensory instability, which may lead to falls and, consequently, fractures or other injuries. For patients with diseases, conditions, or medications that could exacerbate these effects, complete fall risk assessments when initiating antipsychotic treatment and recurrently for patients on long-term antipsychotic therapy.
Seizures: Like other antipsychotic drugs, REXULTI may cause seizures. This risk is greatest in patients with a history of seizures or with conditions that lower the seizure threshold. Conditions that lower the seizure threshold may be more prevalent in older patients.
Body Temperature Regulation: Atypical antipsychotics may disrupt the body's ability to reduce core body temperature. Strenuous exercise, exposure to extreme heat, dehydration, and anticholinergic medications may contribute to an elevation in core body temperature; use REXULTI with caution in patients who may experience these conditions.
Dysphagia: Esophageal dysmotility and aspiration have been associated with antipsychotic drug use. Aspiration pneumonia is a common cause of morbidity and mortality in elderly patients, in particular those with advanced Alzheimer's dementia. Brexpiprazole and other antipsychotic drugs should be used cautiously in patients at risk for aspiration pneumonia.
Pathological Gambling and Other Compulsive Behaviours: Post-marketing case reports suggest that patients can experience intense urges, particularly for gambling, and the inability to control these urges while taking REXULTI. Other compulsive urges, reported less frequently, include: sexual urges, shopping, eating or binge eating, and other impulsive or compulsive behaviours. Because patients may not recognize these behaviours as abnormal, it is important for prescribers to ask patients or their caregivers specifically about the development of new or intense gambling urges, compulsive sexual urges, compulsive shopping, binge or compulsive eating, or other urges while being treated with REXULTI. In some cases, although not all, urges were reported to have stopped when the dose was reduced, or the medication was discontinued. Compulsive behaviours may result in harm to the patient and others if not recognized. Consider dose reduction or stopping the medication if a patient develops such urges.
Lactose: REXULTI film-coated tablets contain lactose. Patients with rare hereditary problems of galactose intolerance, total lactase deficiency or glucose-galactose malabsorption should not take this medicinal product.
Potential for Cognitive and Motor Impairment: REXULTI, like other antipsychotics has the potential to impair judgment, thinking or motor skills. In two 6-week, placebo-controlled clinical trials in patients with MDD, somnolence (including sedation and hypersomnia) was reported in 4% for REXULTI+ADT-treated patients compared to 1% of placebo+ADT patients Studies 1 (Study 228) and 2 (Study 227).
In two 6-week, placebo-controlled clinical trials in patients with schizophrenia, somnolence (including sedation and hypersomnia) was reported in 5% of REXULTI-treated patients compared to 3% of placebo-treated patients Studies 6 (Study 231) and 7 (Study 230).
Patients should be cautioned about operating hazardous machinery including motor vehicles until they are reasonably certain that REXULTI therapy does not affect them adversely.
Special Population: Hepatic Impairment: Patients with moderate to severe hepatic impairment (Child-Pugh score ≥7) generally had higher exposure to brexpiprazole than patients with normal hepatic function; therefore, the maximum recommended dosage is 2 mg once daily for patients with MDD, and 3 mg once daily for patients with schizophrenia. In subjects with varying degrees of hepatic impairment (Child-Pugh Classes A, B, and C), the AUC of oral brexpiprazole (2 mg single dose), compared to matched healthy subjects, increased 24% in mild hepatic impairment, increased 60% in moderate hepatic impairment, and did not change in severe hepatic impairment.
Renal Impairment: Patients with impaired renal function (CLcr<60 mL/minute) had higher exposure to brexpiprazole than patients with normal renal function; therefore, the maximum recommended dosage is 2 mg once daily for patients with MDD, and 3 mg once daily for patients with schizophrenia . In subjects with severe renal impairment (CLcr <30 mL/min), AUC of oral brexpiprazole (2 mg single dose) compared to matched healthy subjects was increased by 68% while its C
max was not changed.
Other Special Populations: No dosage adjustment for brexpiprazole is required on the basis of a patient's sex, race, or smoking status (see Pharmacology: Pharmacokinetics: Special Population under Actions).
Use in Children: Safety and effectiveness in pediatric patients have not been established. Antidepressants increased the risk of suicidal thoughts and behaviours in paediatric patients (See Precautions).
Use in Elderly: Clinical studies of brexpiprazole did not include sufficient numbers of subjects aged 65 and older to determine whether they respond differently from younger subjects. In long-term trials, no new safety concerns were identified in patients 65 years of age or older with MDD (N=132) treated with 1-3 mg of REXULTI as adjunctive treatment to continued antidepressant therapy during 26 weeks In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Based on the results of a safety, tolerability and pharmacokinetics (PK) trial, once daily oral administration of brexpiprazole (up to 3 mg/day for 14 days) as an adjunct therapy in the treatment of elderly subjects (70 to 85 years old, N=11) with MDD were comparable to that of the adult subjects with MDD.
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death compared to placebo. REXULTI is not approved for the treatment of patients with dementia related psychosis.




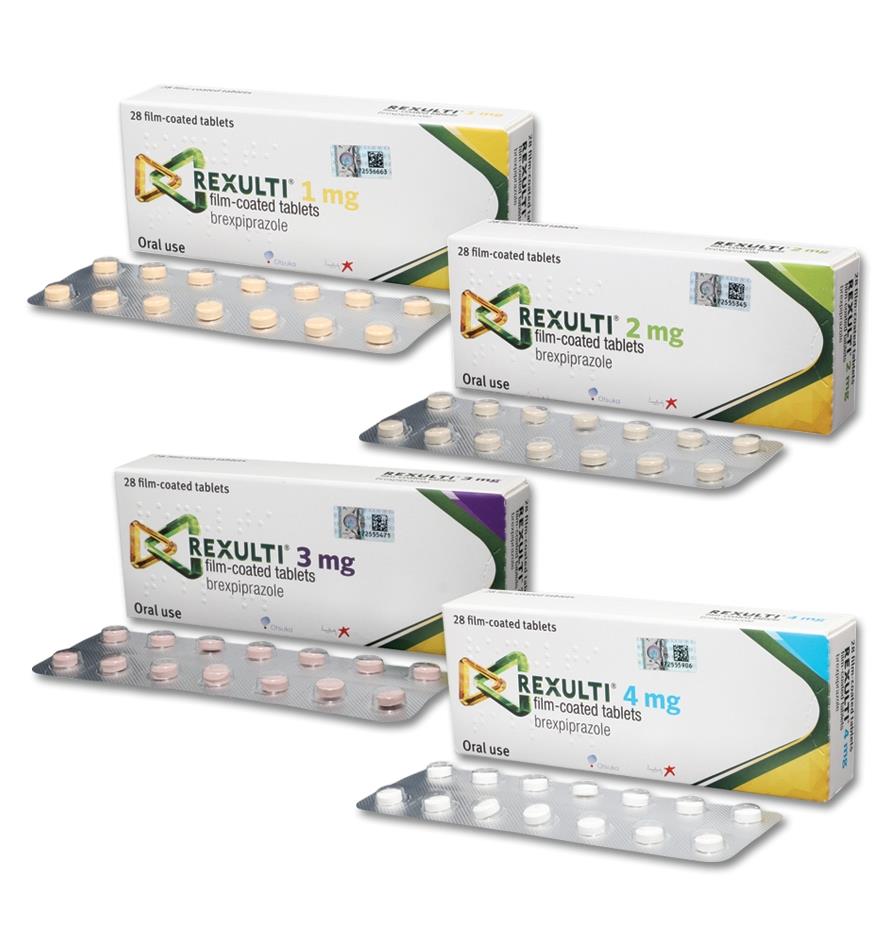
 Click on icon to see table/diagram/image
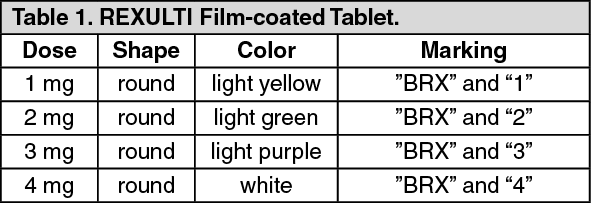
Click on icon to see table/diagram/image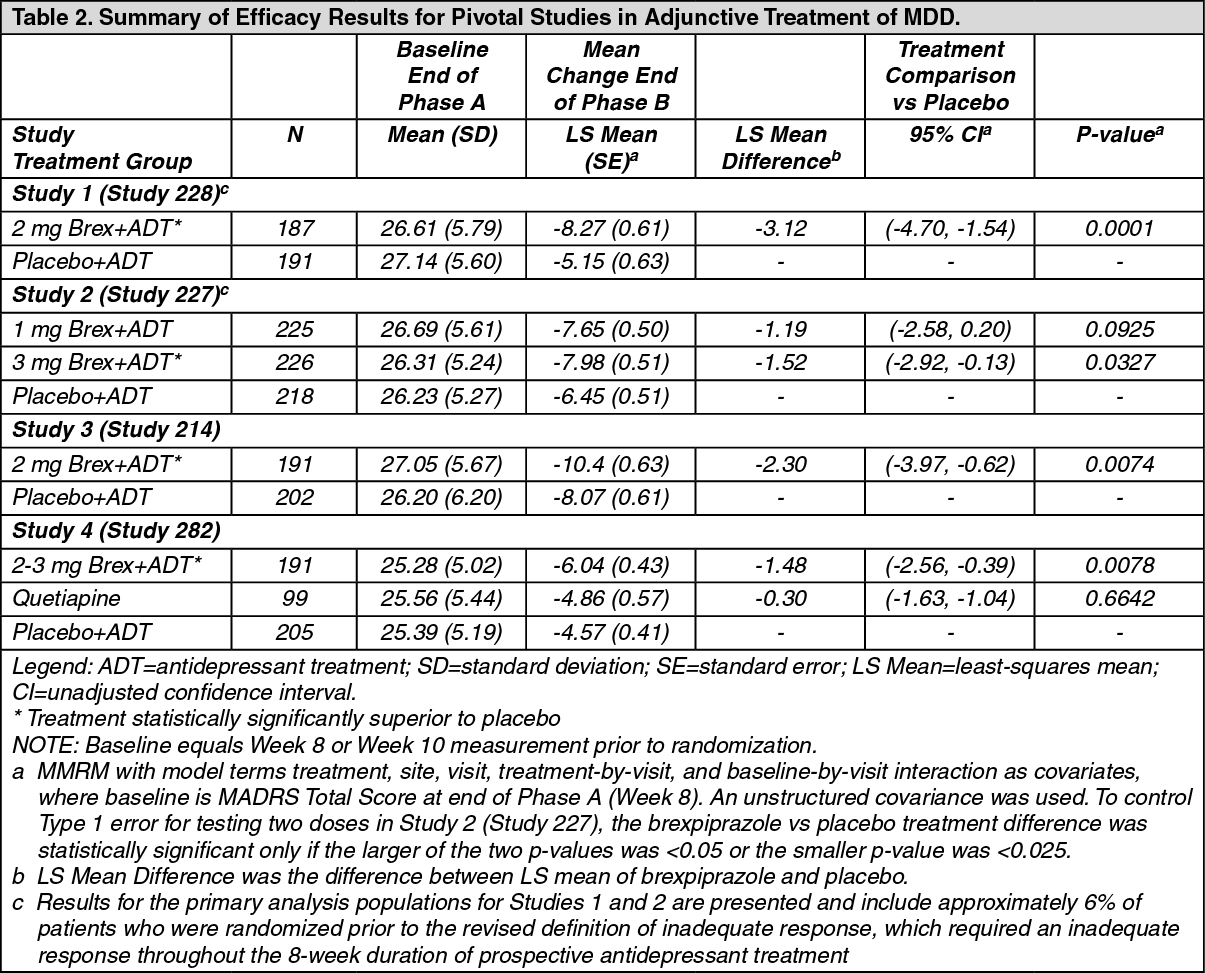 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image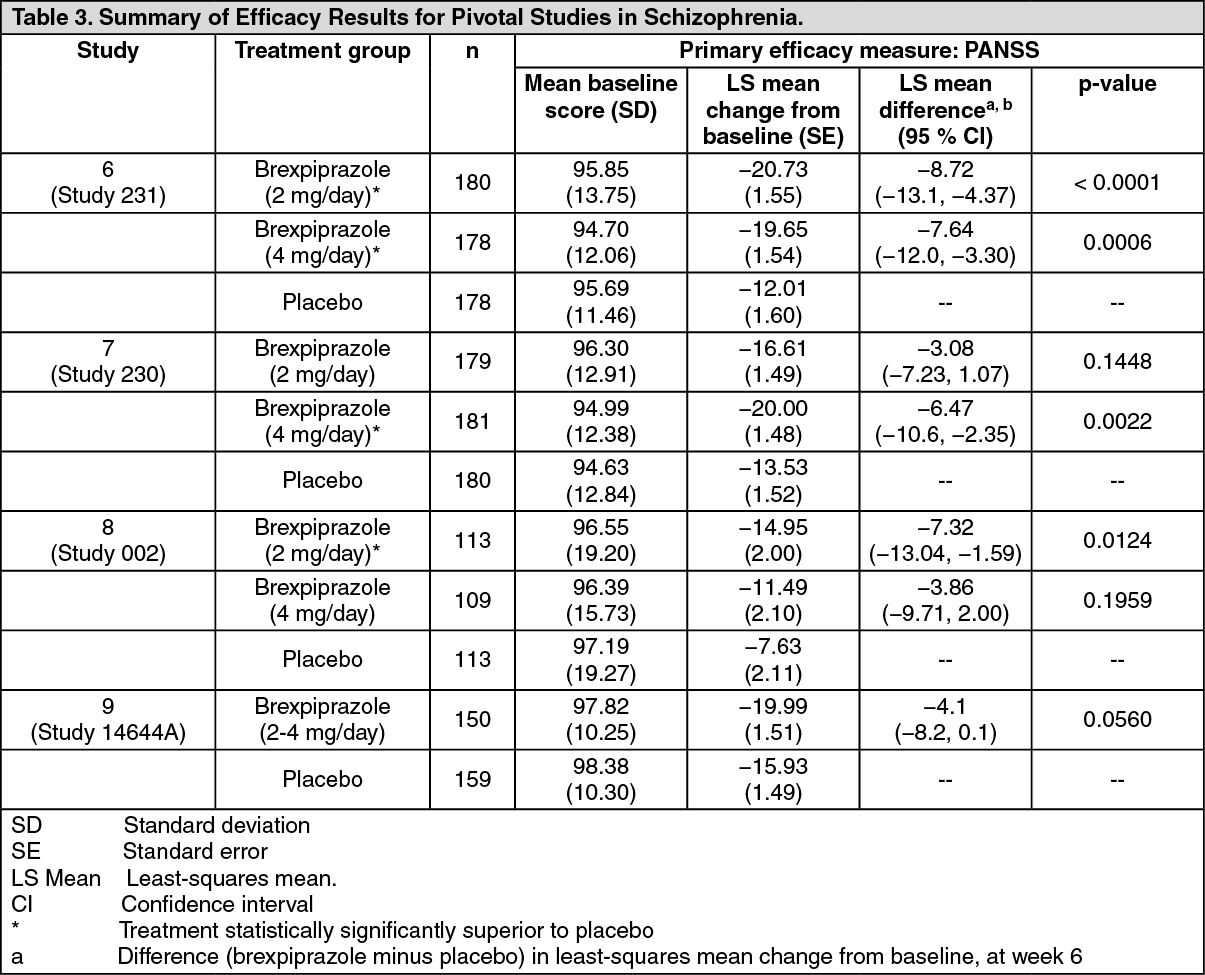 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image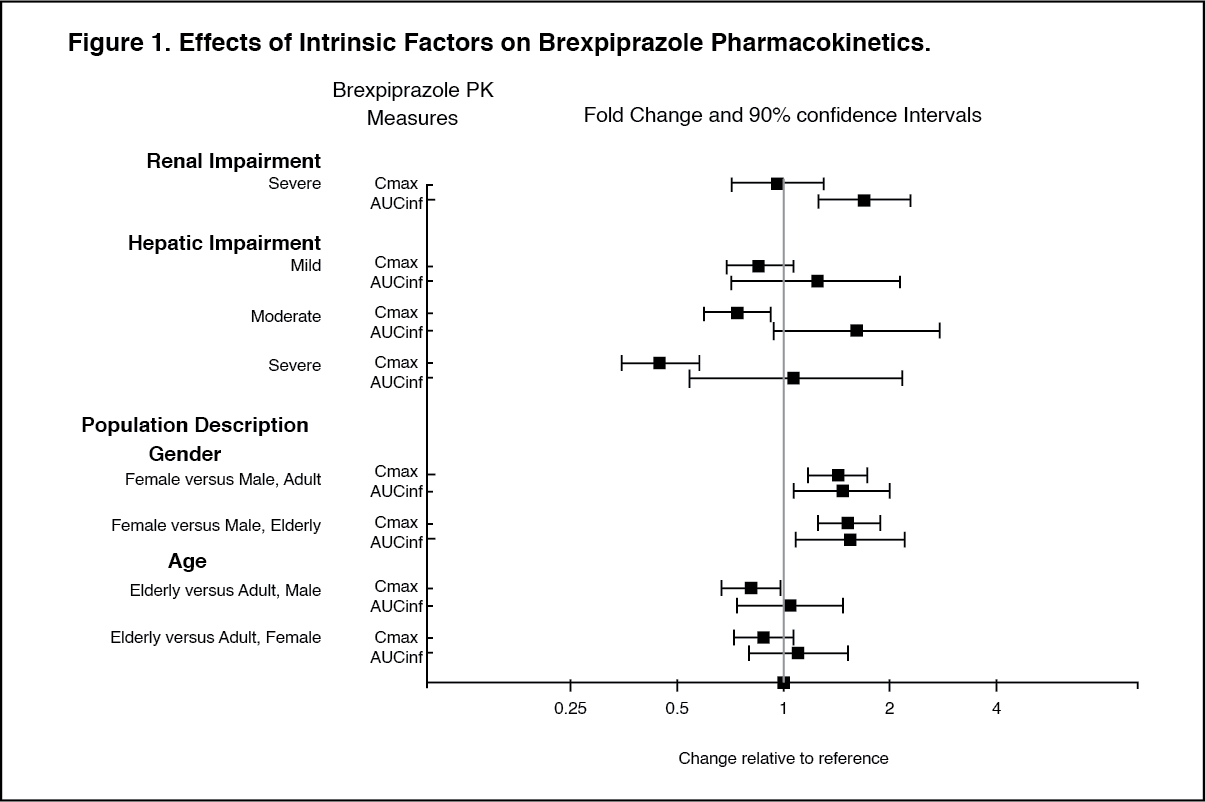 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image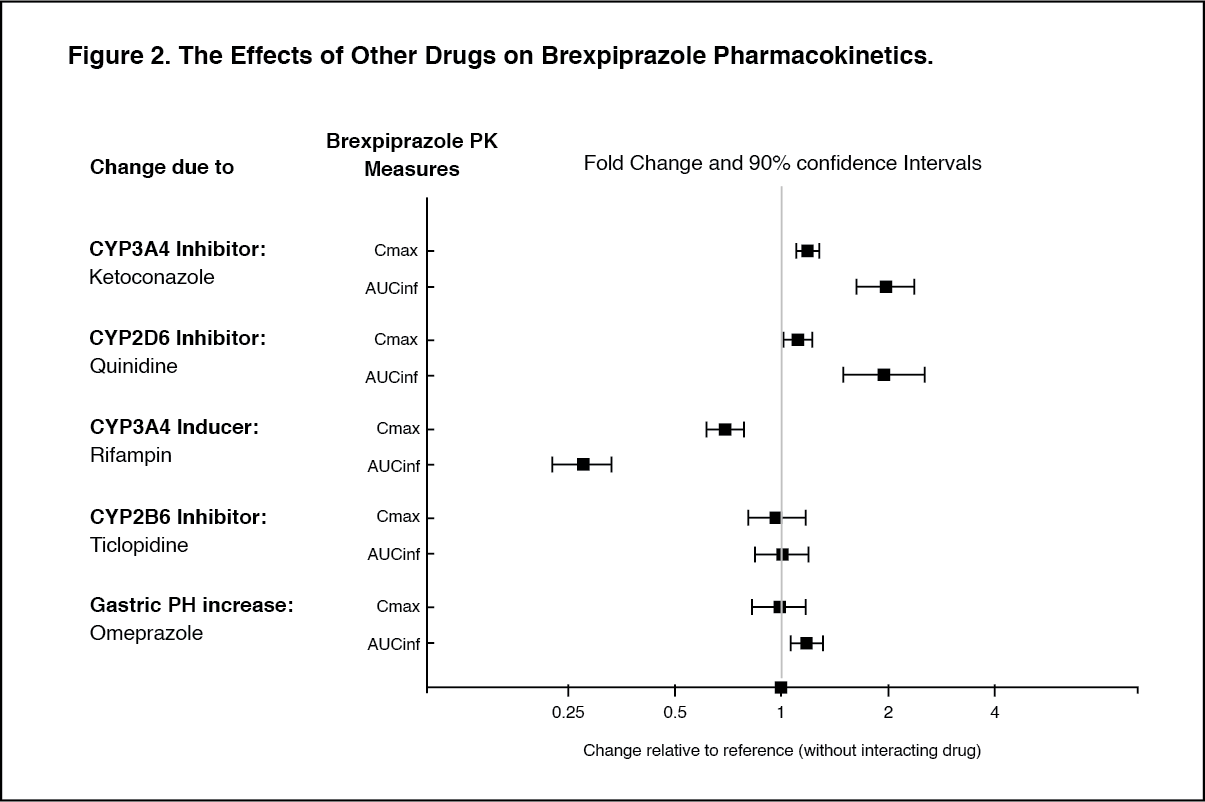 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image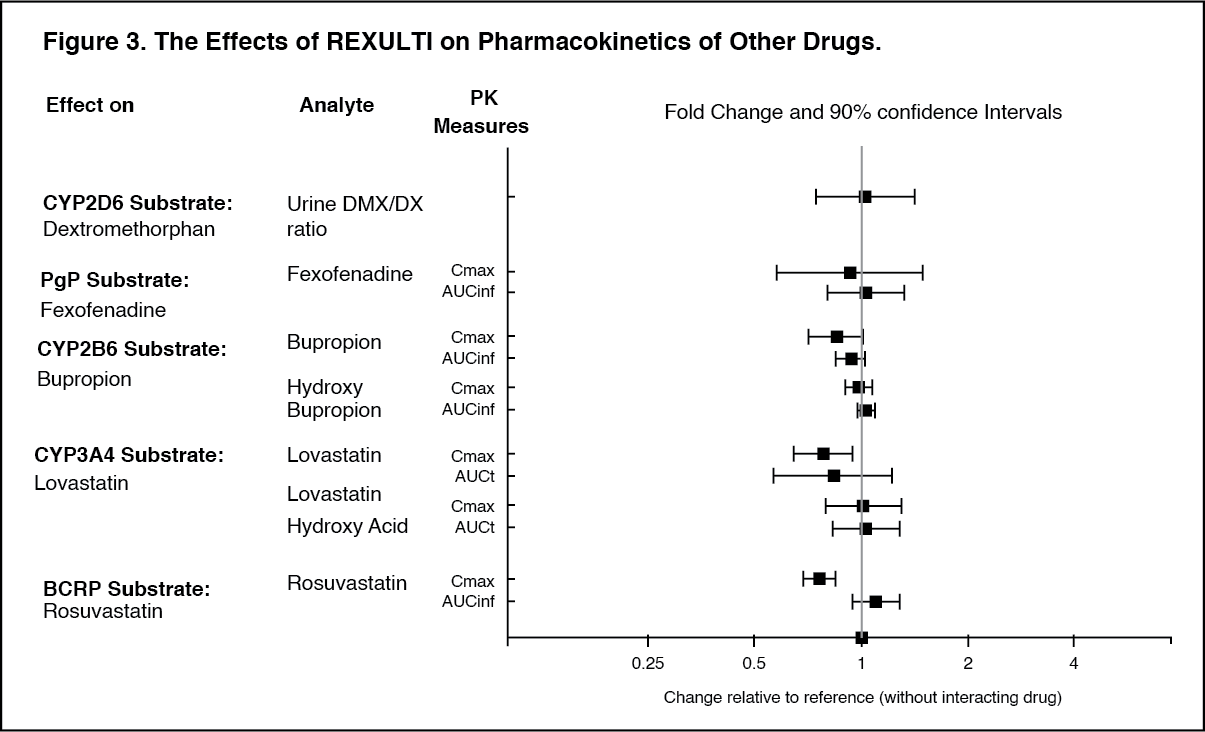 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image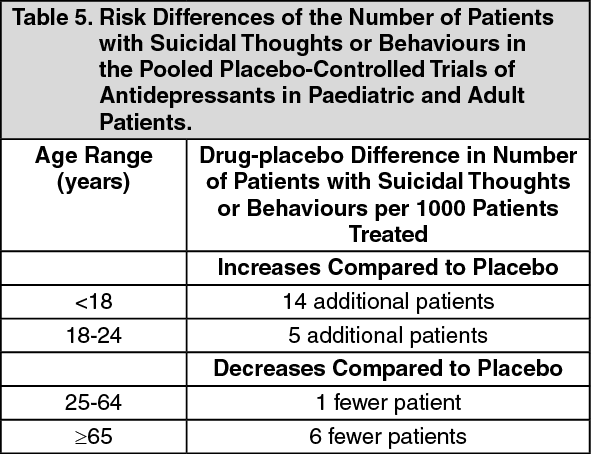 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image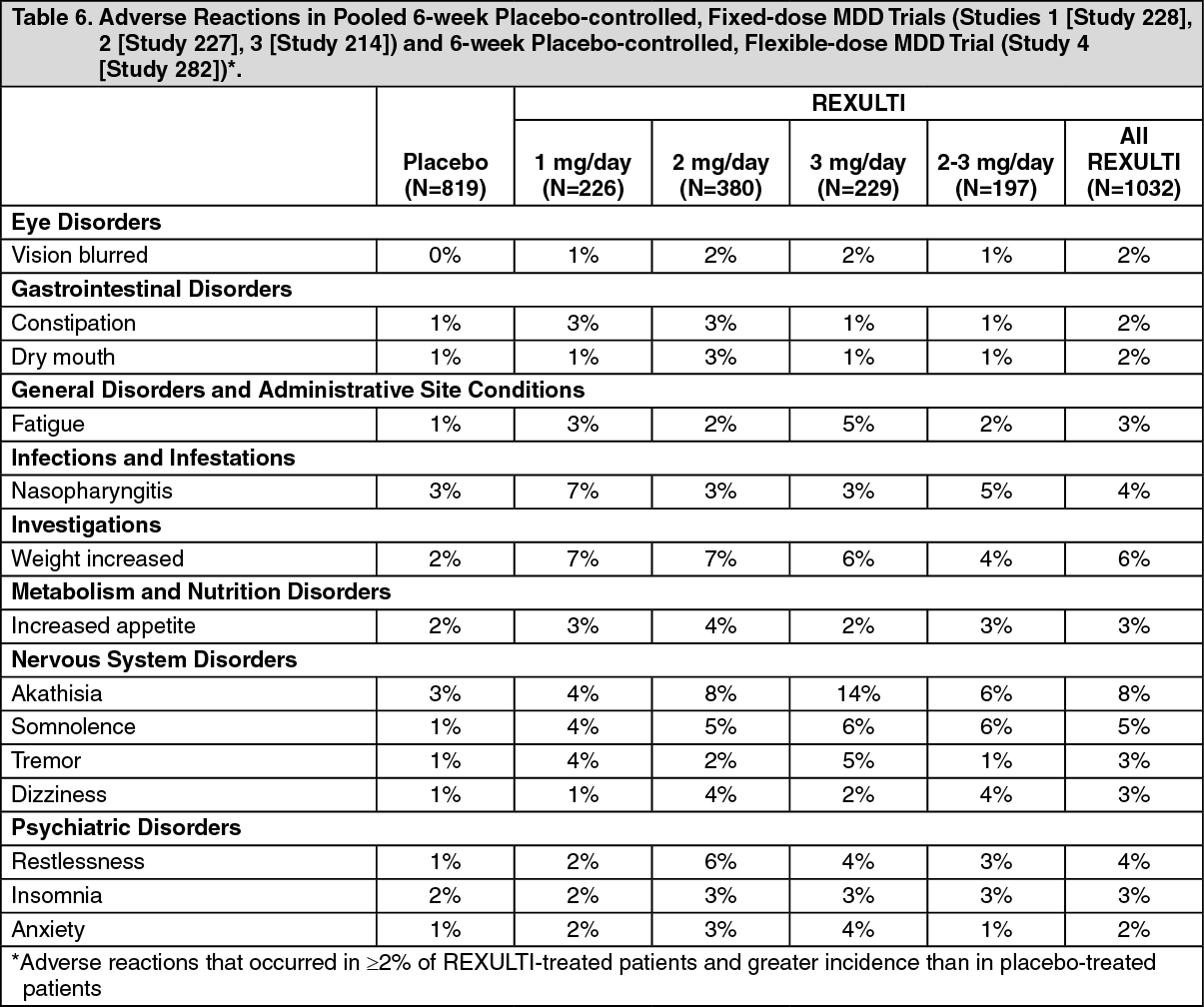 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image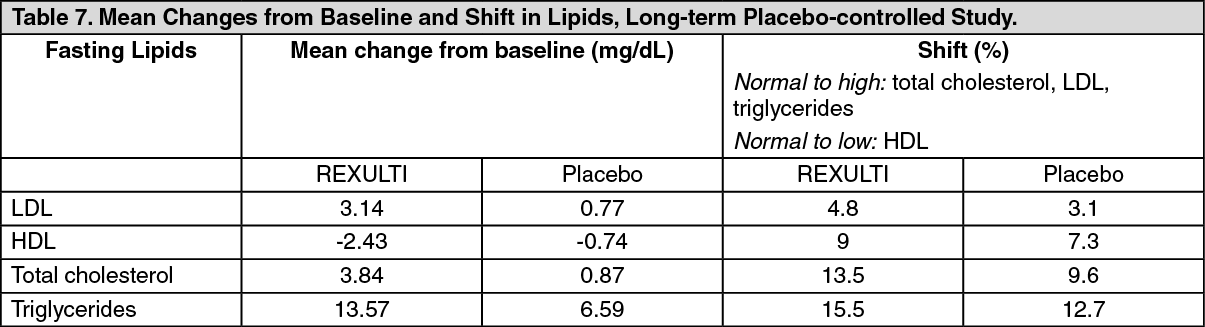 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image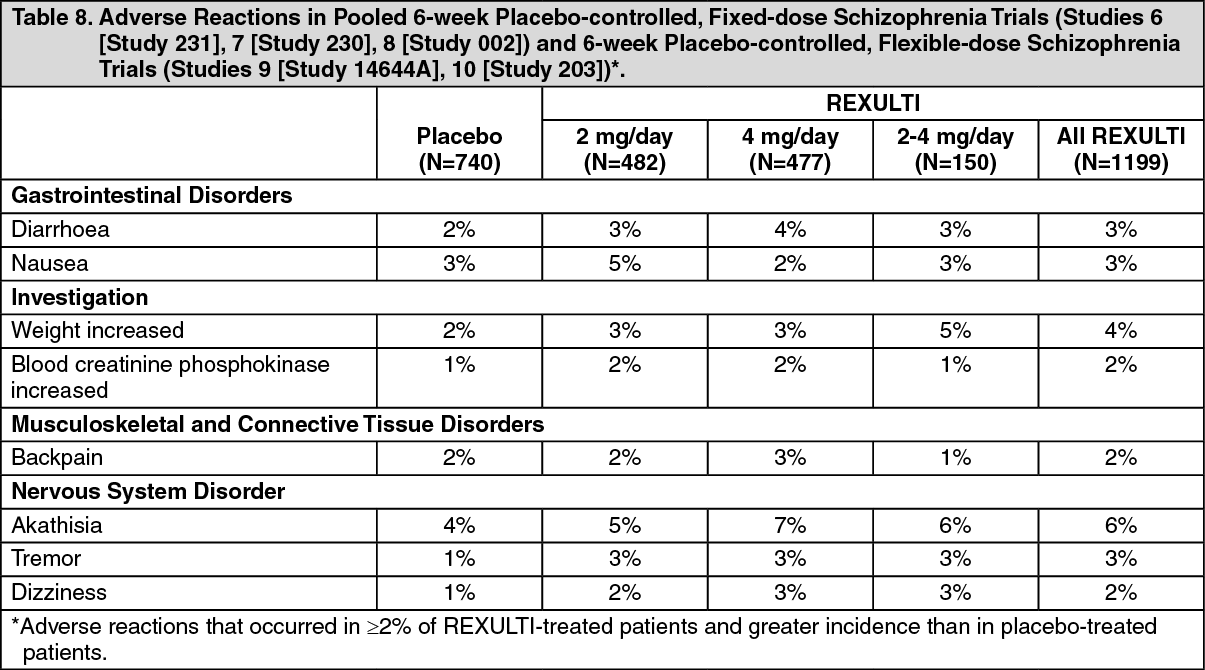 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
