Pharmacotherapeutic group: Nucleoside analogue.
ATC Code: JO5A F05.
Pharmacology: Pharmacodynamics: ZEFFIX is an antiviral agent which is highly active against hepatitis B virus in all cell lines tested and in experimentally infected animals.
Lamivudine is metabolised by both infected and uninfected cells to the triphosphate (TP) derivative which is the active form of the parent compound. The intracellular half-life of the triphosphate in hepatocytes is 17 to 19 hours
in vitro. Lamivudine-TP acts as a substrate for the HBV viral polymerase. The formation of further viral DNA is blocked by incorporation of lamivudine-TP into the chain and subsequent chain termination.
Lamivudine-TP does not interfere with normal cellular deoxynucleotide metabolism. It is also only a weak inhibitor of mammalian DNA polymerases alpha and beta. Furthermore, lamivudine-TP has little effect on mammalian cell DNA content.
In assays relating to potential drug effects on mitochondrial structure and DNA content and function, lamivudine lacked appreciable toxic effects. It has a very low potential to decrease mitochondrial DNA content, is not permanently incorporated into mitochondrial DNA, and does not act as an inhibitor of mitochondrial DNA polymerase γ.
Clinical Studies: ZEFFIX has potent anti-viral activity
in vivo, rapidly suppressing HBV replication following initiation of treatment, resulting in continued HBV suppression, normalisation of serum aminotransferase, significant reductions in liver necro-inflammatory activity, reduced progression of fibrosis and increased HBeAg seroconversion. ZEFFIX has been administered to chronic hepatitis B patients for up to four years in clinical studies. Similar results have been seen in patients regardless of ethnic origin.
In controlled studies in over 800 HBeAg positive patients, treatment with ZEFFIX for one year significantly suppressed HBV DNA replication (34-57% of patients), normalised ALT levels (40 to 72% of patients), induced HBeAg seroconversion (HBeAg and HBV DNA loss with HBeAb detection, 16 to 18% of patients), improved histology (38 to 52% of patients), and reduced progression of fibrosis (3 to 17% of patients) and progression to cirrhosis (1.8% of patients).
The HBeAg seroconversion was maintained in 81% (34/42) of patients off drug followed for approximately 2 years. In addition, HBsAg seroconversion was achieved in 21% (9/42) patients.
In HBeAg positive patients who had not experienced HBeAg seroconversion in one-year controlled studies and were subsequently treated with 2 years of ZEFFIX, 77/128 (60%) had improvement in liver inflammation and 26/51 (51%) had improvement in bridging fibrosis.
In an additional study, after four years of ZEFFIX therapy HBeAg seroconversion (HBeAg loss and HBeAb detection) was seen in 47% (27/58) patients (59% [24/41] of patients with abnormal baseline ALT).
In patients who have not HBeAg seroconverted during treatment, discontinuation of ZEFFIX results in a return of HBV replication with both HBV DNA and serum aminotransferases returning towards pre-treatment levels within 2 to 6 months.
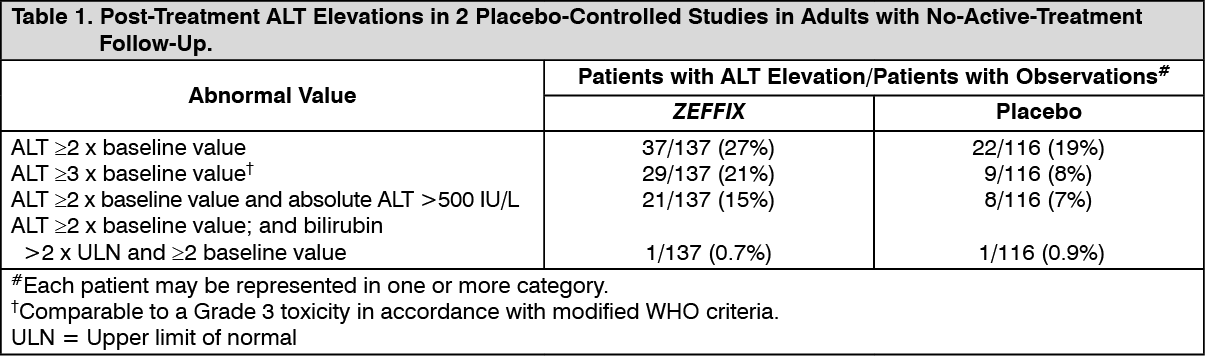
In patients followed for up to 16 weeks after discontinuation of treatment, post-treatment ALT elevations were observed more frequently in patients who had received ZEFFIX than in patients who had received placebo. A comparison of ALT elevations between weeks 52 and 68 in patients who discontinued ZEFFIX at week 52 and patients in the same studies who received placebo throughout the treatment course is shown in Table 1 (see Table 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In a placebo-controlled study of 286 hepatitis B patients aged 2 to 17 years, patients treated with ZEFFIX for one year had a significantly better complete virological response (loss of HBeAg and HBV DNA) compared with placebo (23% [44/191] vs. 13% [12/95]). Normalisation of serum ALT was more frequent in patients treated with ZEFFIX compared with placebo (55% [100/183] vs. 13% [11/88]). In a stratified follow-on study for six months, complete virological response was maintained in 83% [33/40] of patients who had responded after one year of treatment with ZEFFIX and then stopped therapy. ZEFFIX treated patients who did not respond after one year continued treatment for a further 6 months resulting in an additional 10% (12/123) of patients achieving complete virological response and a cumulative complete virological response of 28% (45/163) over 18 months.
HBV viral sub-populations with reduced susceptibility to ZEFFIX
in vitro have been identified. These HBV variants (YMDD variant HBV) are also found in hepatitis B patients who experience a return of detectable serum HBV DNA levels whilst on ZEFFIX treatment. The incidence of YMDD variant HBV (see Precautions), as detected by polymerase chain reaction, increases with duration of treatment; 20% after one year, 53% after three years,70% after four years and may be higher in immunocompromised patients.
Despite the emergence of YMDD variant HBV, patients treated for one year had significantly lower serum HBV DNA and ALT levels and improved liver histology compared to patients on placebo. After 2 years of ZEFFIX treatment, the majority of patients with YMDD variant HBV maintained serum HBV DNA and ALT levels lower than their pre-treatment values, and a proportion experienced HBeAg seroconversion. The adverse event profile is similar for patients with or without YMDD variant HBV.
Given the risk of YMDD mutant HBV, maintenance of lamivudine monotherapy is not appropriate in patients with detectable serum HBV DNA at or beyond 24 weeks of treatment (see Precautions).
In patients with HBeAg negative chronic hepatitis B, the efficacy of ZEFFIX was similar to those infected with wild type HBV (e.g. 71% of patients with HBV DNA suppression, 67% with ALT normalisation and 38% with Knodell HAI-score improvement at one year on treatment). If therapy with ZEFFIX is stopped after one year of treatment, the majority of patients with HBeAg negative chronic hepatitis B have a return of viral replication. Limited data indicate that extended ZEFFIX treatment (two years) maintains HBV DNA suppression and ALT normalisation in this patient population. The incidence of serious adverse events at anytime during and post-treatment was low and similar in patients with HBeAg negative chronic hepatitis B with or without YMDD variant HBV.
In non-controlled studies in liver transplant patients in which ZEFFIX was administered prior to and during transplantation, effective HBV DNA suppression and ALT normalisation was demonstrated. When ZEFFIX therapy was continued post-transplantation there was reduced graft re-infection by HBV, increased HBsAg loss, and a one year survival rate of 76 to 100%. These studies were not placebo-controlled as this was regarded inappropriate in patients with decompensated liver disease.
As anticipated due to the concomitant immunosuppression, the rate of emergence of YMDD variant HBV after 52 weeks treatment was higher (36% to 64%) in liver transplant patients compared with immunocompetent chronic hepatitis B patients (14% to 32%). Studies provide evidence however that the emergence of YMDD variant is not consistently associated with hepatic disease progression and that the majority of patients may continue to benefit from continued ZEFFIX therapy.
Studies of monotherapy with ZEFFIX compared to alpha-interferon alone or in combination for treatment of chronic hepatitis B patients, showed no significant difference in histologic response or HBeAg seroconversion rates between the treatment groups. The safety profile of ZEFFIX was superior to the alpha-interferon containing treatment regimens.
There is no clinical data on the efficacy of ZEFFIX in patients co-infected with Delta hepatitis.
The Antiretroviral Pregnancy Registry has received reports of over 11,000 exposures to lamivudine during pregnancy resulting in live birth, less than 1% of which were in patients with HBV. These consist of over 4,500 exposures during the first trimester, over 7,200 exposures during the second/third trimester and included 143 and 207 major birth defects respectively. The prevalence (95% CI) of defects in the first trimester was 3.1% (2.6, 3.7%) and in the second/third trimester, 2.9% (2.5, 3.3%). Among pregnant women in the reference population, the background rate of birth defects is 2.7%.
Pharmacokinetics: Absorption: Lamivudine is well absorbed from the gastrointestinal tract, and the bioavailability of oral lamivudine in adults is normally between 80 and 85%. Following oral administration, the
mean time (t
max) to maximal serum concentrations (C
max) is about an hour. At therapeutic dose levels i.e. 100 mg once daily, C
max is in the order of 1.1 to 1.5 micrograms/ml and trough levels were 0.015 to 0.020 micrograms/ml.
Co-administration of ZEFFIX with food resulted in a delay of t
max and a lower C
max (decreased by up to 47%). However, the extent (based on the AUC) of lamivudine absorbed was not influenced, therefore ZEFFIX can be administered with or without food.
Distribution: From i.v. studies the mean volume of distribution is 1.3 L/kg. Lamivudine exhibits linear pharmacokinetics over the therapeutic dose range and displays low plasma protein binding to albumin.
Limited data shows lamivudine penetrates the central nervous system and reaches the cerebrospinal fluid (CSF). The mean lamivudine CSF/serum concentration ratio 2-4 hours after oral administration was approximately 0.12.
Metabolism: Lamivudine is predominately cleared by renal excretion of unchanged drug. The likelihood of metabolic drug interactions with lamivudine is low due to the small (5 to 10%) extent of hepatic metabolism and the low plasma protein binding.
Effect of other agents on the pharmacokinetics of lamivudine: Lamivudine is a substrate of MATE1, MATE2-K and OCT2
in vitro. Trimethoprim (an inhibitor of these drug transporters) when given in combination with sulphamethoxazole, has been shown to increase lamivudine plasma concentrations (see Interactions).
Lamivudine is a substrate of the hepatic uptake transporter OCT1. As hepatic elimination plays a minor role in the clearance of lamivudine, drug interactions due to inhibition of OCT1 are unlikely to be of clinical significance.
Lamivudine is an
in vitro substrate of Pgp and BCRP, however due to its high bioavailability it is unlikely that these transporters play a significant role in the absorption of lamivudine. Therefore co-administration of drugs that are inhibitors of these efflux transporters is unlikely to affect the disposition and elimination of lamivudine.
Effect of lamivudine on the pharmacokinetics of other agents:
In vitro, lamivudine demonstrates no or weak inhibition of the drug transporters organic anion transporter 1B1 (OATP1B1), OATP1B3, breast cancer resistance protein (BCRP) or P-glycoprotein (Pgp), multidrug and toxin extrusion protein 1 (MATE1), MATE2-K or organic cation transporter 3 (OCT3). Lamivudine is therefore not expected to affect the plasma concentrations of drugs that are substrates of these drug transporters.
Lamivudine is an inhibitor of OCT1 and OCT2
in vitro with IC50 values of 17 and 33 uM, respectively, however lamivudine has low potential to affect the plasma concentrations of OCT1 and OCT2 substrates at therapeutic drug exposures (up to 300 mg which is three times higher than the recommended maximum dose for HBV).
Elimination: The mean systemic clearance of lamivudine is approximately 0.3 L/h/kg. The observed half-life of elimination is 18 to 19 hours. The majority of lamivudine is excreted unchanged in the urine via glomerular filtration and active secretion (organic cationic transport system).
Renal clearance accounts for about 70% of lamivudine elimination.
Special Patient Populations: Elderly: In elderly patients the pharmacokinetic profile of lamivudine suggests that normal ageing with accompanying renal decline has no clinically significant effect on lamivudine exposure, except in patients with creatinine clearance of less than 50 ml/min (see Dosage & Administration).
Renal impairment: Studies in patients with renal impairment show lamivudine elimination is affected by renal dysfunction. Dose reduction in patients with a creatinine clearance of less than 50 ml/min is necessary (see Dosage & Administration).
Hepatic impairment: A study in hepatically impaired patients (non-HIV and non-HBV infected) showed ZEFFIX is well tolerated in this patient group with no changes in laboratory parameters or the adverse event profile of ZEFFIX. The pharmacokinetics of lamivudine are unaffected by hepatic impairment.
Limited data in patients undergoing liver transplantation show that impairment of hepatic function does not impact significantly on the pharmacokinetics of lamivudine unless accompanied by renal dysfunction.
Pregnancy: Following oral administration, lamivudine pharmacokinetics in late pregnancy were similar to non-pregnant adults.
Lamivudine concentrations in infant serum at birth were similar to those in maternal and cord serum at delivery.
Toxicology: Non-Clinical Information: Administration of ZEFFIX in animal toxicity studies at high doses was not associated with any major organ toxicity. At the highest dosage levels, minor effects on indicators of liver and kidney function were seen together with occasional reduction in liver weights. Reduction of erythrocytes and neutrophil counts were identified as the effects most likely to be of clinical relevance. These events were seen infrequently in clinical studies.
Lamivudine was not mutagenic in bacterial tests but, like many nucleoside analogues showed activity in an
in vitro cytogenetic assay and the mouse lymphoma assay.
Lamivudine was not genotoxic
in vivo at doses that gave plasma concentrations around 60 to 70 times higher than the anticipated clinical plasma levels. As the
in vitro mutagenic activity of ZEFFIX could not be confirmed by
in vivo tests, it is concluded that ZEFFIX should not represent a genotoxic hazard to patients undergoing treatment.
The results of long-term carcinogenicity studies with ZEFFIX in rats and mice did not show any carcinogenic potential.
Reproductive studies in animals have not shown evidence of teratogenicity and showed no effect on male or female fertility in rats. Lamivudine produced small increases in early embryonic loss when administered to pregnant rabbits, at exposure levels comparable to those achieved in man. However, there was no evidence of embryonic loss in rats at exposure levels of approximately 60 times the clinical exposure (based on C
max).


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
