Epirubicin hydrochloride.
Epirubicin 50 mg: Orange-Red Lyophilized powder filled in 30 ml clear moulded glass vial USP Type I with 20 mm bromobutyl slotted rubber stopper & sealed with 20 mm red colour flip off seal.
Each vial contains: Epirubicin Hydrochloride 50 mg.
Pharmacotherapeutic group: Anthracyclines and related substances.
Pharmacology: Pharmacodynamics: The mechanism of action of epirubicin hydrochloride is related to its ability to bind to DNA. Cell culture studies have shown rapid cell penetration, localisation in the nucleus and inhibition of nucleic acid synthesis and mitosis. Epirubicin hydrochloride has proved to be active on a wide spectrum of experimental tumours including L1210 and P388 leukaemias, sarcomas SA180 (solid and ascitic forms), B16 melanoma, mammary carcinoma, Lewis lung carcinoma and colon carcinoma 38. It has also shown activity against human tumours transplanted into athymic nude mice (melanoma, mammary, lung, prostatic and ovarian carcinomas).
Pharmacokinetics: In patients with normal hepatic and renal function, plasma levels after I.V. injection of 60-150 mg/m2 of the drug follow a tri-exponential decreasing pattern with a very fast first phase and a slow terminal phase with a mean half-life of about 40 hours. These doses are within the limits of pharmacokinetic linearity both in terms of plasma clearance values and metabolic pathway. The major metabolites that have been identified are epirubicinol (13-OH epirubicin) and glucuronides of epirubicin hydrochloride and epirubicinol.
The 4'-O-glucuronidation distinguishes epirubicin hydrochloride from doxorubicin and may account for the faster elimination of epirubicin hydrochloride and its reduced toxicity. Plasma levels of the main metabolite, the 13-OH derivative (epirubicinol) are consistently lower and virtually parallel those of the unchanged drug.
Epirubicin hydrochloride is eliminated mainly through the liver; high plasma clearance values (0.9 l/min) indicate that this slow elimination is due to extensive tissue distribution.
Urinary excretion accounts for approximately 9-10% of the administered dose in 48 hours.
Biliary excretion represents the major route of elimination, about 40% of the administered dose being recovered in the bile in 72 hours. The drug does not cross the blood brain barrier.
Epirubicin Hydrochloride has produced responses in a wide range of neoplastic conditions including breast, ovarian, gastric, lung and colorectal carcinomas, malignant lymphomas, leukaemias and multiple myeloma.
Intravesical administration of Epirubicin hydrochloride has been found to be beneficial in the treatment of superficial bladder cancer, carcinoma-in-situ prophylaxis of recurrences after transurethral resection, superficial bladder cancer and advanced metastatic soft tissue sarcoma.
This medicinal product is for intravenous or intravesical use only.
Intravenous administration: It is advisable to administer epirubicin hydrochloride via the tubing of a freely-running IV saline or glucose infusion after checking that the needle is well placed in the vein. Care should be taken to avoid extravasation. In case of extravasation, administration should be stopped immediately.
Conventional dose: When Epirubicin hydrochloride is used as a single agent, the recommended dosage in adults is 60-90 mg/m
2 body area. The drug should be injected I.V. over 3-5 minutes and depending on the patent's haematomedullar status, the dose should be repeated at 21-day intervals.
High doses: Epirubicin hydrochloride as a single agent for the treatment of lung cancer at high doses should be administered according to the following regimens:
Lung cancer: Small cell lung cancer (previously untreated): 120 mg/m2 day 1, every 3 weeks.
Non-small cell lung cancer (squamous, large cell, and adenocarcinoma previously untreated): 135 mg/m2 day 1 or 45 mg/m2 days 1, 2, 3, every 3 weeks.
If signs of toxicity, including severe neutropenia/neutropenic fever and thrombocytopenia occur (which could persist at day 21), dose modification or postponement of the subsequent dose may be required.
Breast Cancer: In the adjuvant treatment of early breast cancer patients with positive lymph nodes, intravenous doses of epirubicin hydrochloride ranging from 100 mg/m
2 (as a single dose on day 1) to 120 mg/m
2 (in two divided doses on days 1 and 8) every 3-4 weeks, in combination with intravenous cyclophosphamide and 5-fluorouracil and oral tamoxifen, are recommended.
For high dose treatment, the drug should be given as an I.V. bolus over 3-5 minutes or as an infusion of up to 30 minutes duration.
Lower doses (60-75 mg/m
2 for conventional treatment and 105-120 mg/m
2 for high dose schedules) are recommended for patients whose bone marrow function has already been impaired by previous chemotherapy or radiotherapy, by age, or neoplastic bone marrow infiltration. The total dose per cycle may be divided over 2-3 successive days.
Combination therapy: When the drug is used in combination with other antitumour agents, the dose needs to be adequately reduced.
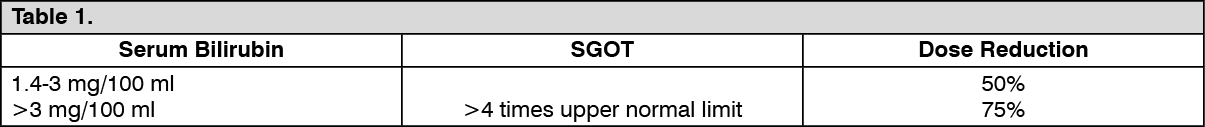
Impaired liver function: Since the major route of elimination of Epirubicin hydrochloride is the hepatobiliary system, the dosage should be reduced in patients with impaired liver function, in order to avoid an increase of overall toxicity based on serum bilirubin levels as follows: See Table 1.
 Click on icon to see table/diagram/image
Renal impairment:
Click on icon to see table/diagram/image
Renal impairment: Moderate renal impairment does not appear to require a dose reduction in view of the limited amount of Epirubicin hydrochloride excreted by this route. However, lower starting doses should be considered in patients with severe renal impairment (serum creatinine >5 mg/dL).
Paediatric population: The safety and efficacy of this medicinal product in children has not been established.
Intravesical administration: Epirubicin hydrochloride can be given by intravesical administration for the treatment of superficial bladder cancer and carcinoma-in-situ. It should not be used in this way for the treatment of invasive tumours which have penetrated the bladder wall where systemic therapy or surgery is more appropriate. Epirubicin hydrochloride has also been successfully used intravesically as a prophylactic agent after transurethral resection of superficial tumours in order to prevent recurrences.
While many regimens have been used, the following may be helpful as a guide: For therapy, 8 x weekly instillations of 50 mg/50 mL (diluted with saline or distilled sterile water).
In the case of local toxicity (chemical cystitis), a dose reduction to 30 mg/50 mL is advised.
For carcinoma-in-situ, depending on the individual tolerability of the patient, the dose may be increased up to 80 mg/50 mL.
For prophylaxis, 4 weekly administrations of 50 mg/50 mL, followed by 11 x monthly instillations at the same dosage, is the schedule most commonly used.
Acute overdosage with epirubicin will result in severe myelosuppression within 10-14 days (mainly leucopenia and thrombocytopenia), gastrointestinal toxic effects (mainly mucosal inflammation) and acute cardiac complications (acute myocardial degeneration within 24 hours). During this period a blood transfusion is required as well as isolation in a sterile room. Latent cardiac failure has been observed with anthracyclines several months (up to 6 months) to years after completion of treatment. Patients must be carefully monitored. If signs of cardiac failure occur, patients should be treated according to conventional guidelines.
Treatment: Symptomatic.
Epirubicin cannot be removed by dialysis.
Hypersensitivity to epirubicin hydrochloride or to any of the excipients, other anthracyclines or anthracenediones.
Lactation.
Intravenous use: patients with marked or persistent myelosuppression induced by previous treatment with either other anti-neoplastic agents or radiotherapy, patients with severe hepatic impairment, severe myocardial insufficiency, recent myocardial infarction, severe arrhythmias, previous treatments with maximum cumulative doses of epirubicin and/or other anthracyclines such as doxorubicin or daunorubicin and anthracenediones, or previous history of cardiac impairment (including 4th degree muscular heart failure, acute heart attack and previous heart attack which led to 3rd and 4th degree muscular heart failure, acute inflammatory heart diseases, severe arrhythmia, myocardiopathy, recent myocardial infarction) patients with acute systemic infections, patients with unstable angina pectoris, myocardiopathy.
Intravesical use: urinary tract infections, inflammation of the bladder, haematuria, invasive tumours penetrating the bladder, catheterisation problems, large volume of residual urine, contracted bladder.
General: Epirubicin should be administered only under the supervision of qualified physicians experienced in the use of cytotoxic therapy.
Diagnostic and treatment facilities should be readily available management of therapy and possible complications due to myelosuppression, especially following treatment with higher doses of epirubicin.
Epirubicin is not active when given orally and should not be injected intramuscularly or intrathecally.
Careful baseline monitoring of various laboratory parameters and cardiac function should precede initial treatment with epirubicin.
Patients should recover from acute toxicities (such as stomatitis, neutropenia, thrombocytopenia, and generalised infections) of prior cytotoxic treatment before beginning treatment with epirubicin.
While treatment with high doses of epirubicin (e.g., ≥90 mg/m2 every 3 to 4 weeks) causes adverse events generally similar to those seen at standard doses (<90 mg/m2 every 3 to 4 weeks), the severity of the neutropenia and stomatitis/mucosal inflammation may be increased. Treatment with high doses of epirubicin does require special attention for possible clinical complications due to profound myelosuppression.
Cardiac Function: Cardiotoxicity is a risk of anthracycline treatment that may be manifested by early (i.e. acute) or late (i.e. delayed) events.
Early (i.e. Acute) Events: Early cardiotoxicity of epirubicin consists mainly of sinus tachycardia and/or electrocardiogram (ECG) abnormalities such as non-specific ST-T wave changes. Tachyarrhythmias, including premature ventricular contractions, ventricular tachycardia, and bradycardia, as well as atrioventricular and bundle-branch block have also been reported. These effects do not usually predict subsequent development of delayed cardiotoxicity, are rarely of clinical importance, and are generally not a consideration for the discontinuation of epirubicin treatment.
Late (i.e. Delayed) Events: Delayed cardiotoxicity usually develops late in the course of therapy with epirubicin or within 2 to 3 months after treatment termination, but later events (several months to years after completion of treatment) have also been reported. Delayed cardiomyopathy is manifested by reduced left ventricular ejection fraction (LVEF) and/or signs and symptoms of congestive heart failure (CHF) such as dyspnoea, pulmonary oedema, dependent oedema, cardiomegaly and hepatomegaly, oliguria, ascites, pleural effusion, and gallop rhythm. Life-threatening CHF is the most severe form of anthracycline-induced cardiomyopathy and represents the cumulative dose-limiting toxicity of the drug.
The risk of developing CHF increases rapidly with increasing total cumulative doses of epirubicin in excess of 900 mg/m2; this cumulative dose should only be exceeded with extreme caution.
Above this level the risk of irreversible congestive heart failure increases greatly.
Heart failure may appear several weeks after discontinuing therapy with epirubicin and may be unresponsive to specific medical treatment. In establishing the maximal cumulative dose of epirubicin, consideration should be given to any concomitant therapy with potentially cardiotoxic drugs.
Cardiac function should be assessed before patients undergo treatment with epirubicin and must be monitored throughout therapy to minimize the risk of incurring severe cardiac impairment. The risk may be decreased through regular monitoring of LVEF during the course of treatment with prompt discontinuation of epirubicin at the first sign of impaired function. The appropriate quantitative method for repeated assessment of cardiac function (evaluation of LVEF) includes multi-gated radionuclide angiography (MUGA) or echocardiography (ECHO). A baseline cardiac evaluation with an ECG and either a MUGA scan or an ECHO is recommended, especially in patients with risk factors for increased cardiotoxicity. Repeated MUGA or ECHO determinations of LVEF should be performed, particularly with higher, cumulative anthracycline doses. The technique used for assessment should be consistent throughout follow-up.
Given the risk of cardiomyopathy, a cumulative dose of 900 mg/m2 epirubicin should be exceeded only with extreme caution.
An ECG is recommended before and after each treatment cycle. Alterations in the ECG tracing, such as flattening or inversion of the Twave, depression of the S-T segment, or the onset of arrhythmias, generally transient and reversible, need not necessarily be taken as indications to discontinue treatment. With cumulative doses <900 mg/m2, there is evidence that cardiac toxicity rarely occurs. In case of cardiac insufficiency, treatment with epirubicin should be discontinued.
Cardiomyopathy induced by anthracyclines is associated with persistent reduction of the QRS voltage, prolongation beyond normal limits of the systolic interval (PEP/LVET) and a reduction of the ejection fraction. Cardiac monitoring of patients receiving epirubicin treatment is highly important and it is advisable to assess cardiac function by non-invasive techniques. Electrocardiogram (ECG) changes may be indicative of anthracycline-induced cardiomyopathy, but ECG is not a sensitive or specific method for following anthracycline-related cardiotoxicity.
Cardiac function monitoring must be particularly strict in patients receiving high cumulative doses and in those with risk factors, particularly prior anthracycline or anthracenedione use. However cardiotoxicity with epirubicin may occur at lower cumulative doses whether or not risk factors are present. It is probable that the toxicity of epirubicin and other anthracyclines or anthracenediones is additive.
Risk factors for cardiac toxicity include active or dormant cardiovascular disease, prior or concomitant radiotherapy to the mediastinal/pericardial area, previous therapy with other anthracyclines or anthracenediones, concomitant use of other drugs with the ability to suppress cardiac contractility or cardiotoxic drugs (e.g. trastuzumab) with an increased risk in the elderly.
Trastuzumab and anthracyclines such as epirubicin should not be used currently in combination except in a well-controlled clinical trial setting with cardiac monitoring. Patients who have previously received anthracyclines are also at risk of cardiotoxicity with trastuzumab treatment, although the risk is lower than with concurrent use of traztuzumab and anthracyclines.
Because the reported half-life of trastuzumab is approximately 28-38 days, trastuzumab may persist in the circulation for up to 27 weeks after stopping trastuzumab treatment. Patients who receive anthracyclines such as epirubicin after stopping trastuzumab may possibly be at increased risk of cardiotoxicity. If possible, physicians should avoid anthracycline-based therapy for up to 27 weeks after stopping trastuzumab. If anthracyclines such as epirubicin are used, the patient's cardiac function should be monitored carefully.
If symptomatic cardiac failure develops during trastuzumab therapy after epirubicin therapy, it should be treated with the standard medications for this purpose.
Haematologic Toxicity: As with other cytotoxic agents, epirubicin may produce myelosuppression. Haematological profiles should be assessed before and during each cycle of therapy with epirubicin. Red blood cell, differential white blood cell (WBC), neutrophil and platelet counts should be carefully monitored both before and during each cycle of therapy. A dose-dependent, reversible leucopenia and/or granulocytopenia (neutropenia) is the predominant manifestation of epirubicin haematologic toxicity and is the most common acute dose-limiting toxicity of this drug. Leukopenia and neutropenia are generally more severe with high-dose schedules, reaching the nadir in most cases between days 10 and 14 after drug administration; this is usually transient with the WBC/neutrophil counts returning to normal values in most cases by day 21. Thrombocytopenia and anaemia may also occur. Clinical consequences of severe myelosuppression include pyrexia, infection, sepsis/septicaemia, septic shock, haemorrhage, tissue hypoxia, or death.
Secondary Leukaemia: Secondary leukaemia, with or without a preleukaemic phase, has been reported in patients treated with anthracyclines, including epirubicin. Secondary leukaemia is more common when such drugs are given in combination with DNA-damaging antineoplastic agents, in combination with radiation treatment, when patients have been heavily pre-treated with cytotoxic drugs, or when doses of the anthracyclines have been escalated. These leukaemia's can have a 1- to 3-year latency period.
Gastrointestinal: Epirubicin is emetigenic. Mucosal inflammation/stomatitis generally appears early after drug administration and, if severe, may progress over a few days to mucosal ulcerations. Most patients recover from this adverse event by the third week of therapy.
Liver Function: The major route of elimination of epirubicin is the hepatobiliary system. Before starting therapy with epirubicin, and during treatment, liver function should be evaluated (SGOT, SGT, alkaline phosphatase, bilirubin, AST). Patients with elevated bilirubin or AST may experience slower clearance of drug with an increase in overall toxicity. Lower doses are recommended in these patients. Patients with severe hepatic impairment should not receive epirubicin.
Renal Function: Serum creatinine should be assessed before and during therapy. Dosage adjustment is necessary in patients with serum creatinine >5 mg/mL.
Epirubicin may impart a red colour to the urine for one or two days after administration.
Effects at Site of Injection: Phlebosclerosis may result from an injection into a small vessel or from repeated injections into the same vein. Following the recommended administration procedures may minimise the risk of phlebitis/thrombophlebitis at the injection site.
Extravasation: Extravasation of epirubicin during intravenous injection may produce local pain, severe tissue lesions (vesication, severe cellulitis) and necrosis. Venous sclerosis may result from injection into small vessels or repeated injections into the same vein. Should signs or symptoms of extravasation occur during intravenous administration of epirubicin, the drug infusion should be immediately discontinued. The adverse effect of extravasation of anthracyclines may be prevented or reduced by immediate use of a specific treatment e.g. dexrazoxane (refer to relevant labels for use). The patient's pain may be relieved by cooling down the area and keeping it cool using hyaluronic acid and DMSO. The patient should be monitored closely during the subsequent period of time, as necrosis may occur after several weeks extravasation occurs, a plastic surgeon should be consulted with a view to possible excision.
Other: As with other cytotoxic agents, thrombophlebitis and thromboembolic phenomena, including pulmonary embolism (in some cases fatal), have been coincidentally reported with the use of epirubicin.
Tumour Lysis Syndrome: Epirubicin may induce hyperuricaemia because of the extensive purine catabolism that accompanies rapid drug-induced lysis of neoplastic cells (tumour-lysis syndrome). Blood uric acid levels, potassium, calcium phosphate, and creatinine should be evaluated after initial treatment. Hydration, urine alkalinisation and prophylaxis with allopurinol to prevent hyperuricaemia may minimize potential complications of tumour-lysis syndrome.
Immunosuppressant Effects/Increased Susceptibility to Infections: Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including epirubicin, may result in serious or fatal infections (see section 4.5). Vaccination with a live vaccine should be avoided in patients receiving epirubicin. Killed or inactivated vaccines may be administered; however, the response to such vaccines may be diminished.
Reproductive system: Epirubicin can cause genotoxicity. Men and women treated with epirubicin should adopt appropriate contraceptives. Patients desiring to have children after completion of therapy should be advised to obtain genetic counselling if appropriate.
Additional warnings and precautions for other routes of administration: Intravesical route: Administration of epirubicin may produce symptoms of chemical cystitis (such as dysuria, polyuria, nocturia, stranguria, haematuria, bladder discomfort, necrosis of the bladder wall) and bladder constriction. Special attention is required for catheterization problems (e.g., uretheral obstruction due to massive intravesical tumours).
Intra-arterial route: Intra-arterial administration of epirubicin (transcatheter arterial embolization for the localized or regional therapies of primary hepatocellular carcinoma or liver metastases) may produce (in addition to systemic toxicity qualitatively similar to that observed following intravenous administration of epirubicin) localized or regional events which include gastro-duodenal ulcers (probably due to reflux of the drugs into the gastric artery) and narrowing of bile ducts due to drug induced sclerosing cholangitis. This route of administration can lead to widespread necrosis of the perfused tissue.
Pregnancy: Women of child-bearing potential should be advised to avoid becoming pregnant during treatment and should use effective contraceptive methods.
There is no conclusive information as to whether epirubicin may adversely affect human fertility or cause teratogenesis. Experimental data in animals suggest that epirubicin may cause foetal harm when administered to a pregnant woman. Like most other anti-cancer agents, epirubicin has shown mutagenic and carcinogenic properties in animals. Both men and women receiving epirubicin should be informed of the potential risk of adverse effects on reproduction. Women of childbearing potential should be fully informed of the potential hazard to the foetus. If epirubicin is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the foetus. There are no studies in pregnant women. Epirubicin should be used during pregnancy only if the potential benefit justifies the potential risk to the foetus.
Lactation: It is not known whether epirubicin is excreted in human milk. Because many drugs, including other anthracyclines, are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from epirubicin, mothers should discontinue nursing prior to taking this drug.
The following undesirable effects have been observed and reported during treatment with epirubicin with the following frequencies: More than 10 % of treated patients can expect to develop undesirable effects. The most common undesirable effects are myelosuppression, gastrointestinal side effects, anorexia, alopecia, infection. (See Table 2.)
Click on icon to see table/diagram/image
Description of selected adverse reactions: Neoplasms benign, malignant and unspecified (including cysts and polyps): Secondary acute myeloid leukaemia with or without a pre-leukaemic phase, in patients treated with epirubicin in combination with DNA-damaging antineoplastic agents.
These leukaemias have a short (1-3 years) latency.
Blood and lymphatic system disorders: High doses of epirubicin have been safely administered in a large number of untreated patients having various solid tumours and have caused adverse events which are not different from those seen at conventional doses with the exception of reversible severe neutropenia (<500 neutrophils/mm
3 for <7 days) which occurred in the majority of patients. Only few patients required hospitalisation and supportive therapy for severe infectious complications at high doses.
Skin and subcutaneous tissue disorders: Alopecia, normally reversible, appears in 60-90 % of treated cases; it is accompanied by lack of beard growth in males.
General disorders and administration site conditions: Mucositis may appear 5-10 days after the start of treatment, and usually involves stomatitis with areas of painful erosions, ulceration and bleeding, mainly along the side of the tongue and the sublingual mucosa.
Local pain and tissue necrosis (following accidental paravenous injection) may occur.
Intravesical administration: As only a small amount of active ingredient is reabsorbed after intravesical instillation, severe systemic adverse reactions as well as allergic reactions are rare. Commonly reported are local reactions like burning sensation and frequent voiding (pollakisuria). Occasional bacterial or chemical cystitis have been reported. These adverse reactions are mostly reversible.
Epirubicin is mainly used in combination with other cytotoxic drugs. Additive toxicity may occur especially with regard to bone marrow/hematologic and gastro-intestinal effects. Patients should be monitored for additive toxicity, especially myelotoxicity and gastrointestinal toxicity.
The use of epirubicin in combination chemotherapy with other potentially cardiotoxic drugs, as well as the concomitant use of other cardioactive compounds (e.g., calcium channel blockers), requires monitoring of cardiac function throughout treatment.
Epirubicin is extensively metabolised by the liver. Changes in hepatic function induced by concomitant therapies may affect epirubicin metabolism, pharmacokinetics, therapeutic efficacy and/or toxicity.
Anthracyclines including epirubicin should not be administered in combination with other cardiotoxic agents unless the patient's cardiac function is closely monitored. Patients receiving anthracyclines after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab, may also be at an increased risk of developing cardiotoxicity. The half-life of trastuzumab is approximately 28-38 days and may persist in the circulation for up to 27 weeks. Therefore, physicians should avoid anthracycline-based therapy for up to 27 weeks after stopping trastuzumab when possible. If anthracyclines are used before this time, careful monitoring of cardiac function is recommended.
Vaccination with a live vaccine should be avoided in patients receiving epirubicin. Killed or inactivated vaccines may be administered; however, the response to such vaccines may be diminished.
Drug interactions with epirubicin have been observed with cimetidine, dexverapamil, dexrazoxane, docetaxel, interferon α2B, paclitaxel, trastuzumab and quinine.
Cimetidine 400 mg b.i.d given prior to epirubicin 100 mg/m2 every 3 weeks led to a 50% increase in epirubicin AUC and a 41% increase in epirubicinol AUC (latter p<0.05). The AUC of the 7-eoxydoxorubicinol aglycone and liver blood flow were not reduced, so results are not explained by reduced cytochrome P-450 activity. Cimetidine should be discontinued during treatment with epirubicin.
When given prior to epirubicin, paclitaxel can cause increased plasma concentrations of unchanged epirubicin and its metabolites, the latter being, however, neither toxic nor active.
Co-administration of paclitaxel or docetaxel did not affect the pharmacokinetics of epirubicin when epirubicin was administered prior to the taxane.
This combination may be used if using staggered administration between the two agents. Infusion of epirubicin and paclitaxel should be performed with at least a 24 hour interval between the 2 agents.
Dexverapamil may alter the pharmacokinetics of epirubicin and possibly increase its bone marrow depressant effects.
One study found that docetaxel may increase the plasma concentrations of epirubicin metabolites when administered immediately after epirubicin.
Quinine may accelerate the initial distribution of epirubicin from blood into the tissues and may have an influence on the red blood cells partitioning of epirubicin.
The co-administration of interferon α2b may cause a reduction in both the terminal elimination half-life and the total clearance of epirubicin.
The possibility of a marked disturbance of haematopoiesis needs to be kept in mind with a (pre-) treatment with medications which influence the bone marrow (i.e. cytostatic agents, sulphonamide, chloramphenicol, diphenylhydantoin, amidopyrine-derivate, antiretroviral agents).
Prior administration of higher doses (900 mg/m2 and 1200 mg/m2) of dexrazoxane may increase the systemic clearance of epirubicin and result in a decrease in AUC.
Increase of myelosuppression may occur in patients receiving combination therapy of anthracycline and dexrazoxane.
The potential risk of cardiotoxicity may increase in patients who have received concomitant cardiotoxic agents (e.g. 5-fluorouracil, cyclophosphamide, cisplatin, taxanes), or concomitant (or prior) radiotherapy to the mediastinal area.
Direction for Reconstitution: Reconstitute with 25 mL of Sterile Water for Injection resulting in a solution concentrate of 2 mg/mL shake vigorously. It may take up to 4 minutes to completely dissolve. The reconstituted solution should be used immediately or with in 24 hours, if stored at between 2-8°C.
Instructions and Special Precautions for Handling and Disposal: 1. If an infusion solution is to be prepared, this should be performed by trained personnel under aseptic conditions.
2. Preparation of an infusion solution should be performed in a designated aseptic area.
3. Adequate protective disposable gloves, goggles, gown and mask should be worn.
4. Precautions should be taken to avoid the medicinal product accidentally coming into contact with the eyes. In the event of contact with the eyes, irrigate with large amounts of water and/or 0.9% sodium chloride solution. Then seek medical evaluation by a physician.
5. In case of skin contact, thoroughly wash the affected area with soap and water or sodium bicarbonate solution. However, do not abrade the skin by using a scrub brush. Always wash hands after removing gloves.
6. Spillage or leakage should be treated with dilute sodium hypochlorite (1% available chlorine) solution, preferably by soaking, and then water. All cleaning materials should be disposed of as detailed as follows.
7. Pregnant staff should not handle the cytotoxic preparation.
8. Adequate care and precautions should be taken in the disposal of items (syringes, needles, etc) used to reconstitute and/or dilute cytotoxic medicinal products. Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
Store at temperatures not exceeding 30°C.
L01DB03 - epirubicin ; Belongs to the class of cytotoxic antibiotics, anthracyclines and related substances. Used in the treatment of cancer.
Cytobin-50 lyo powd for inj 50 mg
(glass) 30 mL x 1's



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image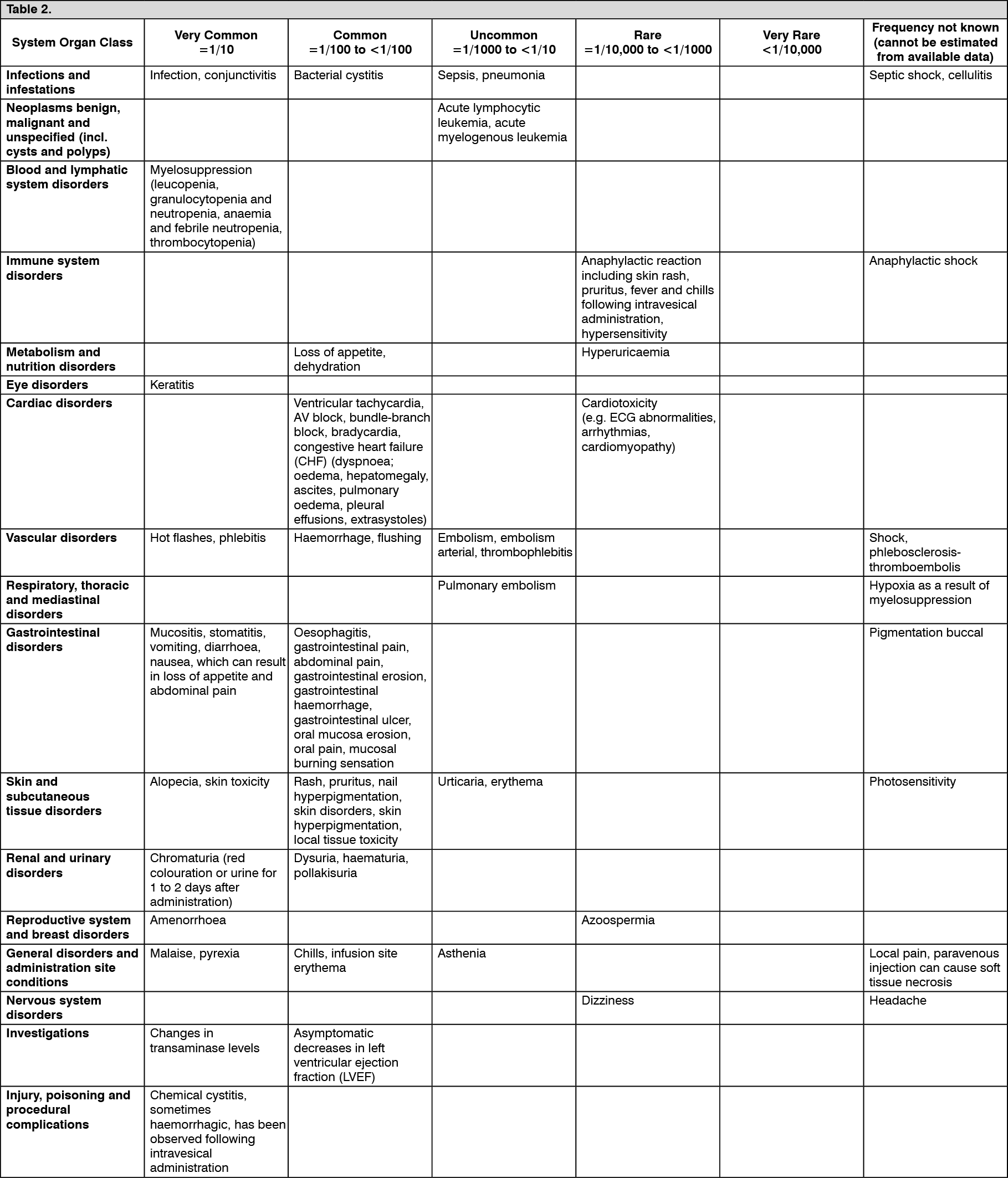 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out