


 Sign Out
Sign Out

Infusion related reactions (with symptoms such as dyspnea, bronchospasm, urticaria, flushing, rash, and increased blood pressure and heart rate) have also been reported in patients treated with vedolizumab.
Vedolizumab has been studied in three placebo-controlled clinical trials in patients with ulcerative colitis (GEMINI I) or Crohn's disease (GEMINI II and III). In two controlled studies (GEMINI I and II) involving 1,434 patients receiving vedolizumab 300 mg at Week 0, Week 2 and then every eight weeks or every four weeks from Week 6 for up to 52 weeks, and 297 patients receiving placebo for up to 52 weeks, adverse events were reported in 84% of vedolizumab-treated patients and 78% of placebo-treated patients. Over 52 weeks, 19% of vedolizumab-treated patients experienced serious adverse events compared to 13% of placebo-treated patients. Similar rates of adverse events were seen in the every eight week and every four week dosing groups in the Phase 3 clinical trials.
The proportion of patients who discontinued treatment due to adverse events was 9% for vedolizumab-treated patients and 10% for placebo-treated patients. In the combined studies of GEMINI I and II the adverse reactions that occurred in ≥5% were nausea, nasopharyngitis, upper respiratory tract infection, arthralgia, pyrexia, fatigue, headache, cough. Infusion-related reactions were reported in 4% of patients receiving vedolizumab.
In the shorter (10 week) placebo controlled induction trial, GEMINI III, the types of adverse reactions reported were similar but occurred at lower frequency than the longer 52 week trials.
A further 279 patients were treated with vedolizumab at Week 0 and Week 2 and then with placebo for up to 52 weeks. Of these patients, 84% experienced adverse events and 15% experienced serious adverse events.
Patients (n=1,822) previously enrolled in Phase 2 or 3 vedolizumab studies were eligible to enroll in an ongoing open-label study and received vedolizumab 300 mg every four weeks.
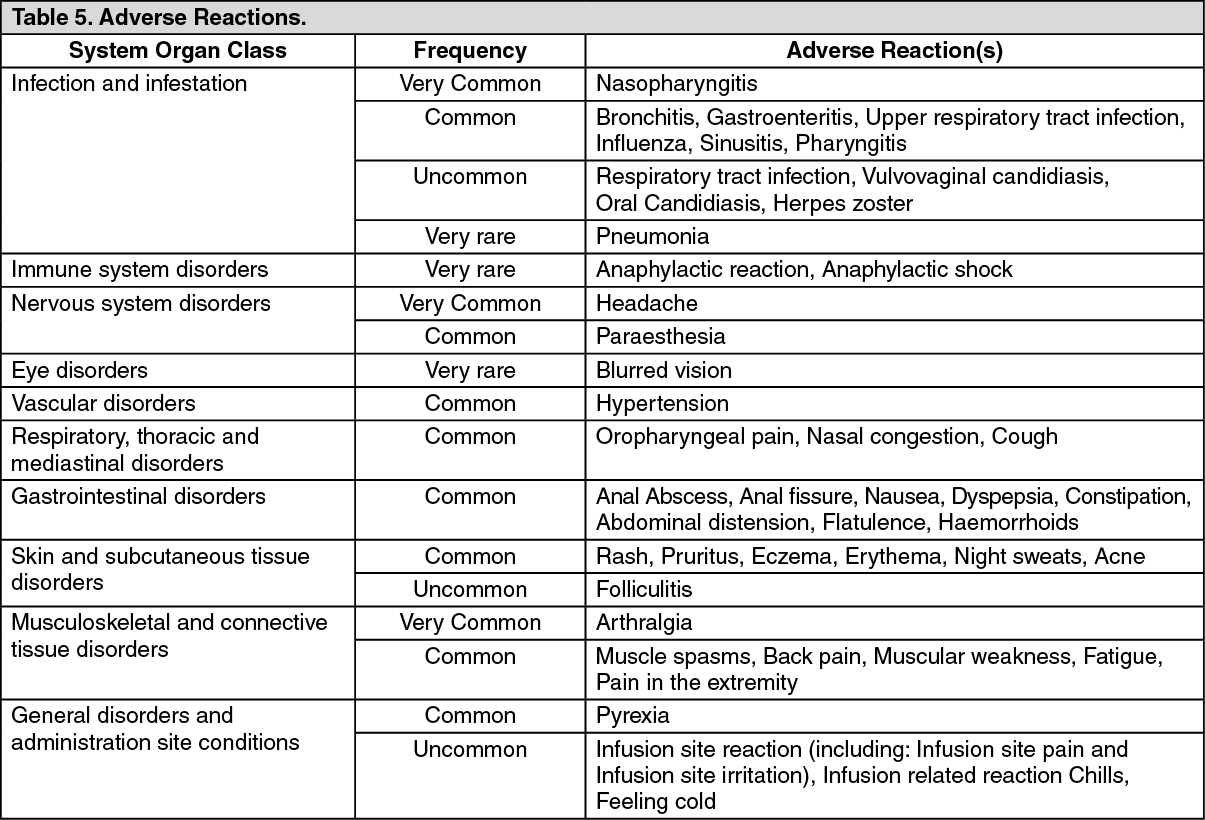
Tabulated list of adverse reactions: The following listing of adverse reactions is based on the clinical trial experience and are displayed by system organ class. Within the system organ classes, adverse reactions are listed under headings of the following frequency categories: very common (≥1/10), common (≥1/100 to <1/10) and uncommon (≥1/1,000 to <1/100), very rare (< 1/10,000) and not known (cannot be estimated from the available data). Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness. (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse reactions: Infusion-related reactions: In GEMINI 1 and 2 controlled studies, 4% of intravenous vedolizumab-treated patients and 3% of placebo-treated patients experienced an adverse event defined by the investigator as infusion-related reaction (IRR) (see Precautions). No individual Preferred Term reported as an IRR occurred at a rate above 1%.
The majority of IRRs were mild or moderate in intensity and <1% resulted in discontinuation of study treatment. Observed IRRs generally resolved with no or minimal intervention following the infusion. Most infusion related reactions occurred within the first 2 hours. Of those patients who had infusion related reactions, those dosed with intravenous vedolizumab had more infusion related reactions with in the first two hours as compared to placebo patients with infusion related reactions. Most infusion related reactions were not serious and occurred during the infusion or within the first hour after infusion is completed.
One serious adverse event of IRR was reported in a Crohn's disease patient during the second infusion (symptoms reported were dyspnoea, bronchospasm, urticaria, flushing, rash, and increased blood pressure and heart rate) and was successfully managed with discontinuation of infusion and treatment with antihistamine and intravenous hydrocortisone. In patients who received vedolizumab at Weeks 0 and 2 followed by placebo, no increase in the rate of IRR was seen upon retreatment with intravenous vedolizumab after loss of response.
Infections: In GEMINI 1 and 2 controlled studies, with intravenous vedolizumab, the rate of infections was 0.85 per patient-year in the vedolizumab-treated patients and 0.70 per patient-year in the placebo-treated patients.
The infections consisted primarily of nasopharyngitis, upper respiratory tract infection, sinusitis, and urinary tract infections. Most patients continued on vedolizumab after the infection resolved.
In GEMINI 1 and 2 controlled studies with intravenous vedolizumab, the rate of serious infections was 0.07 per patient year in vedolizumab-treated patients and 0.06 per patient year in placebo-treated patients.
Over time, there was no significant increase in the rate of serious infections.
In controlled and open-label studies in adults with intravenous vedolizumab, serious infections have been reported, which include tuberculosis, sepsis (some fatal), salmonella sepsis, listeria meningitis, and cytomegaloviral colitis.
In clinical studies with intravenous vedolizumab, the rate of infections in vedolizumab treated patients with BMI of 30 kg/m2 and above was higher than for those with BMI less than 30 kg/m2.
In clinical studies with intravenous vedolizumab, a slightly higher incidence of serious infections was reported in vedolizumab treated patients who had prior exposure to TNFα antagonist therapy compared to patients who were naïve to previous TNFα antagonist therapy.
Malignancy: Overall, results from the clinical program to date do not suggest an increased risk for malignancy with vedolizumab treatment; however, the number of malignancies was small and long-term exposure was limited. Long-term safety evaluations are ongoing.
Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product.
Healthcare professionals are asked to report any suspected adverse reactions to the marketing authorization holder.
The patient must seek medical attention immediately at the first sign of any adverse drug reaction.
View ADR Monitoring Form