There is no data available on undesirable effects of this combination. However, side effects have been reported with individual molecules.
Atorvastatin: In the atorvastatin placebo-controlled clinical trial database of 16,066 patients treated for a mean period of 53 weeks, 5.2% of patients on atorvastatin discontinued due to adverse reactions compared to 4.0% of the patients on placebo.
Based on data from clinical studies and extensive post-marketing experience, the following table presents the adverse reaction profile for Atorvastatin.
Estimated frequencies of reactions are ranked according to the following convention: common (≥ 1/100, < 1/10); uncommon (≥ 1/1,000, < 1/100); rare (≥ 1/10,000, < 1/1,000); very rare (< 1/10,000), not known (cannot be estimated from the available data).
Infections and infestations: Common: nasopharyngitis.
Blood and lymphatic system disorders: Rare: thrombocytopenia.
Immune system disorders: Common: allergic reactions.
Very rare: anaphylaxis.
Metabolism and nutrition disorders: Common: hyperglycaemia.
Uncommon: hypoglycaemia, weight gain, anorexia.
Psychiatric disorders: Uncommon: nightmare, insomnia.
Nervous system disorders: Common: headache.
Uncommon: dizziness, paraesthesia, hypoesthesia, dysgeusia, amnesia.
Rare: peripheral neuropathy.
Eye disorders: Uncommon: blurred vision.
Rare: visual disturbance.
Ear and labyrinth disorders: Uncommon: tinnitus.
Very rare: hearing loss.
Respiratory, thoracic and mediastinal disorders: Common: pharyngolaryngeal pain, epistaxis.
Gastrointestinal disorders: Common: constipation, flatulence, dyspepsia, nausea, diarrhoea.
Uncommon: vomiting, abdominal pain upper and lower, eructation, pancreatitis.
Hepatobiliary disorders: Uncommon: hepatitis.
Rare: cholestasis.
Very rare: hepatic failure.
Skin and subcutaneous tissue disorders: Uncommon: urticaria, skin rash, pruritus, alopecia.
Rare: angioneurotic oedema, dermatitis bullous including erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis.
Musculoskeletal and connective tissue disorders: Common: myalgia, arthralgia, pain in extremity, muscle spasms, joint swelling, back pain.
Uncommon: neck pain, muscle fatigue.
Rare: myopathy, myositis, rhabdomyolysis, tendonopathy sometimes complicated by rupture.
Not known: immune mediated necrotizing myopathy.
Reproductive system and breast disorders: Very rare: gynecomastia.
General disorders and administration site conditions: Uncommon: malaise, asthenia, chest pain, peripheral oedema, fatigue, pyrexia.
Investigations: Common: liver function test abnormal, blood creatine kinase increased.
Uncommon: while blood cells urine positive.
As with other HMG-CoA reductase inhibitors elevated serum transaminases have been reported in patients receiving Atorvastatin. These changes were usually mild, transient, and did not require interruption of treatment. Clinically important (> 3 times upper normal limit) elevations in serum transaminases occurred in 0.8% patients on Atorvastatin. These elevations were dose related and were reversible in all patients.
Elevated serum creatine kinase (CK) levels greater than 3 times upper limit of normal occurred in 2.5% of patients on Atorvastatin, similar to other HMG-CoA reductase inhibitors in clinical trials. Levels above 10 times the normal upper range occurred in 0.4% Atorvastatin treated patients.
The following adverse events have been reported with some statins: Sexual dysfunction.
Depression.
Exceptional cases of interstitial lung disease, especially with long term therapy.
Diabetes Mellitus: Frequency will depend on the presence or absence of risk factors (fasting blood glucose ≥5.6 mmol/L, BMI >30kg/m
2, raised triglycerides, history of hypertension).
Fenofibrate: The most commonly reported ADRs during fenofibrate therapy
are digestive, gastric or intestinal disorders. The following undesirable effects have been observed during placebo-controlled clinical trials (n=2344) and post-marketing with the indicated frequencies as follows: see Table 2.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image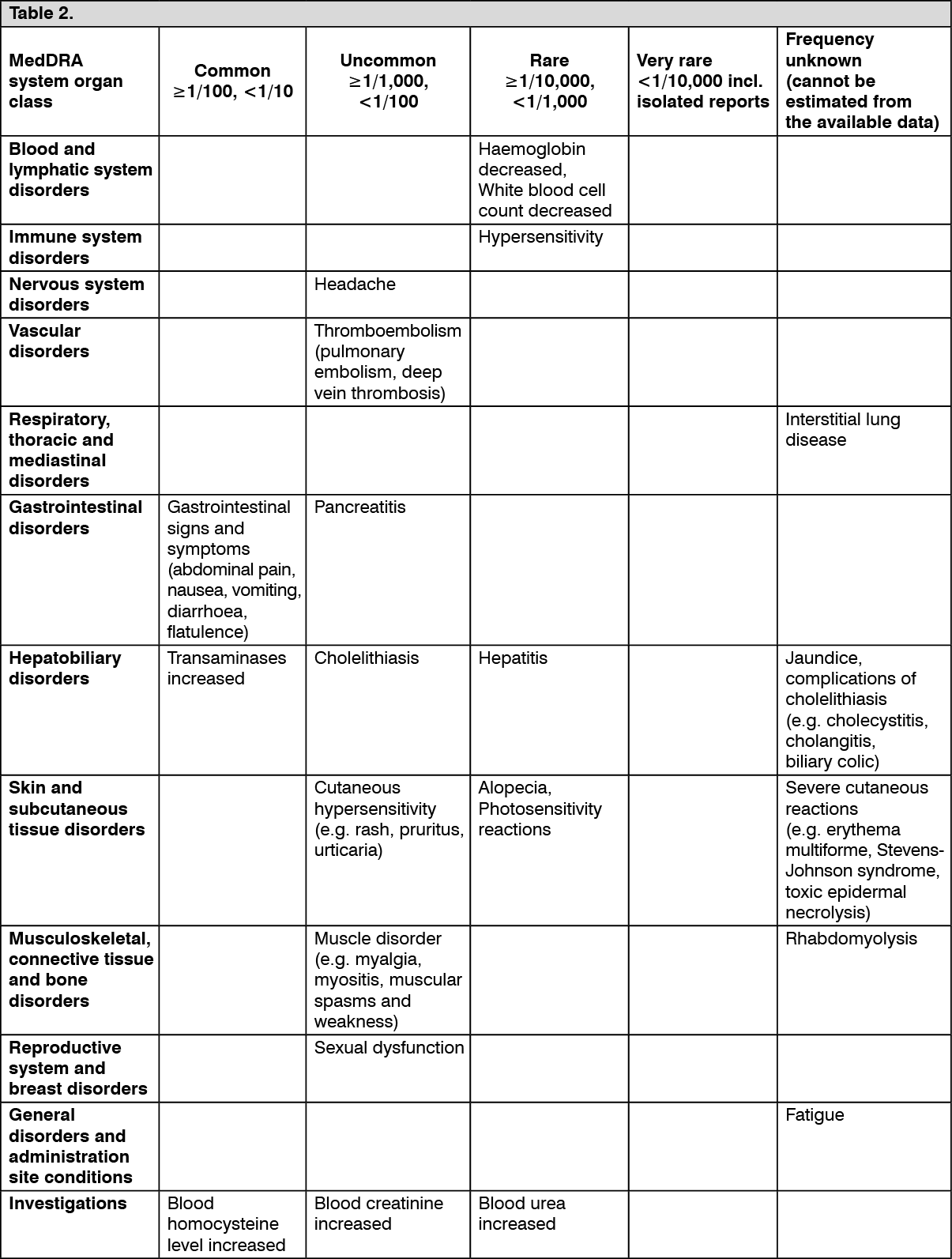 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out