Potassium-Competitive Acid Blocker (PCAB).
Pharmacology: Pharmacodynamics: Pharmacological actions: The Product has a mechanism of action to inhibit gastric acid secretion by controlling H+/K+-ATPase in parietal cells of stomach in a K+ ion-dependent and reversible manner. The Product directly inhibits the proton pump without undergoing acid-induced activity.
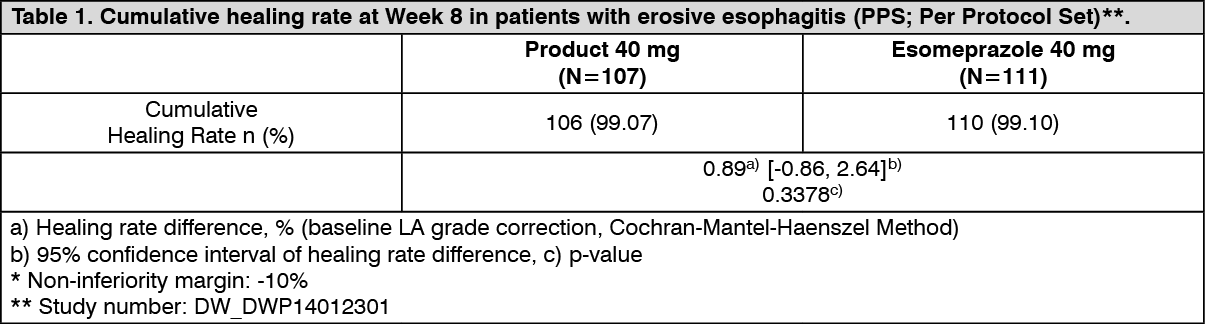
Clinical Studies: Erosive esophagitis: A randomized, double-blind, comparative phase 3 clinical trial with 40 mg of the Product or 40 mg of Esomeprazole administered orally once daily for up to 8 weeks was conducted in 218 patients with erosive esophagitis. As a study result, the cumulative healing rate at Week 8 is shown in the table as follows and the non-inferiority of the Product to esomeprazole group was confirmed (Table 1). (See Table 1.)
 Click on icon to see table/diagram/image
Pharmacokinetics: Absorption:
Click on icon to see table/diagram/image
Pharmacokinetics: Absorption: When single administration of 10-320 mg of the Product carried out in healthy adults, it was rapidly absorbed and the maximum plasma concentration reached at a median value of 1.75-3.5 hours after administration. Blood drug concentration increased as the dose increased. When 20-160 mg of the Product was repeatedly administered orally for 7 days, the drug concentration in the steady state and terminal elimination half-life were similar to that of the single administration. There was no accumulation of in vivo exposure after repeated administration and the drug concentration in blood tended to increase in proportion to the dose increase.
As a result of orally administering 160 mg of the Product to healthy adult males after fasting and high-fat diet to evaluate the dietary effect on bioavailability, there were no significant differences in in vivo exposure and pharmacodynamic endpoints (retention time above pH 4 in the stomach).
Distribution: The in vitro plasma protein binding rates in human plasma were 94.3% and 92.8% at concentrations of 1 and 10 μg/mL, respectively.
Metabolism: The Product is mainly metabolized by CYP3A4, the main metabolite is metabolite M14 and this metabolite is ineffective.
Excretion: The amounts of unchanged forms excreted from urine and feces after intravenous administration in rats were 0.61% and 34.22%. After oral administration of the 14
C marker of the Product to rats, the excretion recovery rate at 120 hours was 98.9%, where the recovery rates of urine and feces were 18.8% and 80.1 %, respectively.
After single oral administration to biliary tract intubated rats, the bile was excreted at 88.0% at 48 hours and the total recovery rate was 98.2%. After oral administration of the 14
C marker of the Product to dogs, the fecal recovery rate at 168 hours was 96.7%, where the recovery rates of urine and feces were 38.8% and 57.9%, respectively.
After oral administration of the Product to healthy adult males, the mean elimination half-life of unchanged forms and metabolite M14 were 9.7 hours and 14.2 hours, respectively. The urine excretions and elimination rate of the unchanged form were about 0.6% and 0.63 L/hr, respectively.
Drug-Drug Interaction: Drugs that may affect the plasma concentration of the Product: The Product is a substrate of CYP3A4 and when the Product and a CYP3A4 inhibitor were administered in combination, the increase in exposure of the Product may be slight.
The AUC
t (Area under the plasma concentration-time curve from time zero to time t) of the Product increased slightly to 1.1 times as a result of co-administration of 80 mg of the Product and 500 mg as clarithromycin twice a day for 7 days in healthy adult males.
Drugs whose plasma concentration may be changed by the Product: The Product showed competitive inhibitory effect against CYP3A4 in in vitro, but its IC
50 (11.7 μM) was about 100 times higher than the maximum plasma concentration in the clinical dose (40 mg basis).
Asa result of co-administration of three preparations, 80 mg of the Product, 1 g as amoxicillin and 500 mg as clarithromycin twice a day for 7 days to healthy adult males, the AUC
t and C
ss,max of clarithromycin were decreased to 23% and 28%, respectively and the AUC
t and C
ss,max of amoxicillin were decreased to 14% and 33%, respectively.
The Product showed competitive inhibitory effect against MATE1, MATE2K and OCT1 in in vitro, but it is unlikely to increase the blood concentration of the transporter substrate drug when considering the maximum plasma concentration at the clinical dose (40 mg basis).
Toxicology: Genotoxicity: The Product was negative in all of the bacterial reverse mutation tests using Salmonella and E. coli, chromosomal abnormality tests using CHO cell lines and micronucleus tests using rats.
Reproductive toxicity: As a result of fertility and early embryo development tests in rats, there was no effect on fertility and early embryo development up to a dose of 50 mg/kg/day.
As a result of the embryo-fetal development test in rats, a decrease in feed intake and weight was observed in the group administered with more than 30 mg/kg/day.
The weight of the fetus decreased by 5%, but there was no effect on development or growth delay. The no-effect levels (NOELs) of embryo-fetal and the maternal were confirmed to be 60 mg/kg/day (about 19.8 times the clinical dose of 40 mg AUC) and 15 mg/kg/day (7.5 times the clinical dose 40 mg AUC), respectively. As a result of the embryo-fetal development test in rabbits, a decrease in feed intake and weight and constipation symptoms were observed in the group administered with more than 15 mg/kg/day. However, there was no effect on development or growth delay. The no-effect levels (NOELs) of the maternal and embryo-fetal were confirmed to be 10 mg/kg/day (0.6 times the clinical dose of 40 mg AUC) and 15 mg/kg/day (1.7 times the clinical dose 40 mg AUC), respectively.
As a result of prenatal development and maternal function evaluation tests in rats, there was no effect on the offspring immediately after childbirth by the Product, but it was transferred to maternal milk and the body weight decreased during the breastfeeding period in the administration dose of more than 7.5 mg/kg/day (3.7 times the clinical dose 40 mg AUC). However, no dysfunctions including behavior, development, sexual maturity and genital organs of the offspring even at the high dose of 30 mg/kg/day (17.2 times the clinical dose 40 mg AUC).
Carcinogenicity: In a carcinogenicity study administered orally in rats for 2 years, gastric neuroendocrine tumors were observed at 10 mg/kg/day (about 3. 7 times based on the clinical dose 40 mg/day AUC) for males and 5 mg/kg/day (about 4.1 times based on the clinical dose 40 mg/day AUC) for females. In the 26-week carcinogenicity test in RasH2 mice, gastric benign adenomas were observed at 60 mg/kg/day in males (about 26.9 times based on the clinical dose 40 mg/day AUC).



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out