Each tablet contains 15 mg or 30 mg of tolvaptan.
Excipients/Inactive Ingredients: Maize starch, Hydroxypropylcellulose, Lactose monohydrate, Magnesium stearate, Microcrystalline cellulose, Indigo carmine aluminium lake.
Pharmacotherapeutic group: Diuretics, vasopressin antagonists.
PHARMACOLOGY: Pharmacodynamics: Mechanism of action: Tolvaptan is a vasopressin antagonist that specifically blocks the binding of arginine vasopressin (AVP) at the V2 receptors of the distal portions of the nephron. Tolvaptan affinity for the human V2 receptor is 1.8 times that of native AVP.
Pharmacodynamic effects: The pharmacodynamic effects of tolvaptan have been determined in healthy subjects and subjects with ADPKD across CKD stages 1 to 4. Effects on free water clearance and urine volume are evident across all CKD stages with smaller absolute effects observed at later stages, consistent with the declining number of fully functioning nephrons. Acute reductions in mean total kidney volume were also observed following 3 weeks of therapy in all CKD stages, ranging from -4.6% for CKD stage 1 to -1.9% for CKD stage 4.
Clinical efficacy and safety: The primary focus of the clinical program for development of tolvaptan tablets for the treatment of ADPKD is a single pivotal, multinational, phase 3, randomised, placebo controlled trial in which the long-term safety and efficacy of oral split dose tolvaptan regimens (titrated between 60 mg/day and 120 mg/day) were compared with placebo in 1,445 adult subjects with ADPKD. In total, 14 clinical trials involving tolvaptan have been completed worldwide in support of the ADPKD indication, including 8 trials in the US, 1 in the Netherlands, 3 in Japan, 1 in Korea, and the multinational phase 3 pivotal trial.
The phase 3 pivotal trial (TEMPO 3:4, 156-04-251) included subjects from 129 centres in the Americas, Japan, Europe and other countries. The primary objective of this trial was to evaluate the long-term efficacy of tolvaptan in ADPKD through rate of total kidney volume (TKV) change (%) for tolvaptan-treated compared with placebo-treated subjects. In this trial a total of 1,445 adult patients (age 18-50 years) with evidence of rapidly-progressing, early ADPKD (meeting modified Ravine criteria, total kidney volume (TKV) ≥750 mL, estimated creatinine clearance ≥60 mL/min) were randomized 2:1 to treatment with tolvaptan or placebo. Patients were treated for up to 3 years.
Tolvaptan (n = 961) and placebo (n = 484) groups were well matched in terms of gender with an average age of 39 years. The inclusion criteria identified patients who at baseline had evidence of early disease progression. At baseline, patients had average estimated glomerular filtration rate (eGFR) of 82 ml/min/1.73 m2 (CKD-EPI) with 79% having hypertension and a mean TKV of 1,692 mL (height adjusted 972 mL/m). Approximately 35% of subjects were chronic kidney disease (CKD) stage 1, 48% CKD stage 2, and 17% CKD stage 3 (eGFRCKD-EPI). While these criteria were useful in enriching the study population with patients who were rapidly progressing, subgroup analyses based on stratification criteria (age, TKV, GFR, Albuminuria, Hypertension) indicated the presence of such risk factors at younger ages predicts more rapid disease progression.
The results of the primary endpoint, the rate of change in TKV for subjects randomised to tolvaptan (normalised as percentage) to the rate of change for subjects on placebo, were highly statistically significant. The rate of TKV increase over 3 years was significantly less for tolvaptan-treated subjects than for subjects receiving placebo: 2.80% per year vs 5.51% per year, respectively (ratio of geometric mean 0.974; 95% CI 0.969 to 0.980; p <0.0001).
Pre-specified secondary endpoints were tested sequentially. The key secondary composite endpoint (ADPKD progression) was time to multiple clinical progression events of: 1) worsening kidney function (defined as a persistent [reproduced over at least 2 weeks] 25% reduction in reciprocal serum creatinine during treatment [from end of titration to last on-medicinal product visit]); 2) medically significant kidney pain (defined as requiring prescribed leave last-resort analgesics, narcotic and anti-nociceptive, radiologic or surgical interventions); 3) worsening hypertension; 4) worsening albuminuria.
The relative rate of ADPKD-related events was decreased by 13.5% in tolvaptan-treated patients, (hazard ratio, 0.87; 95% CI, 0.78 to 0.97; p = 0.0095).
The result of the key secondary composite endpoint is primarily attributed to effects on worsening kidney function and medically significant kidney pain. The renal function events were 61.4% less likely for tolvaptan compared with placebo (hazard ratio, 0.39; 95% CI, 0.26 to 0.57; nominal p < 0.0001), while renal pain events were 35.8% less likely in tolvaptan-treated patients (hazard ratio, 0.64; 95% CI, 0.47 to 0.89; nominal p = 0.007). In contrast, there was no effect of tolvaptan on either progression of hypertension or albuminuria.
TEMPO 4:4 is an open-label extension study that included 871 subjects that completed TEMPO 3:4 from 106 centres across 13 countries. This trial evaluated the effects of tolvaptan on safety, TKV and eGFR in subjects receiving active treatment for 5 years (early-treated), compared with subjects treated with placebo for 3 years, then switched to active treatment for 2 years (delayed-treated).
The primary end point for TKV did not distinguish a difference in change (-1.7%) over the 5 year treatment between early- and delayed-treated subjects at the pre-specified threshold of statistical significance (p = 0.3580). Both groups' TKV growth trajectory was slowed, relative to placebo in the first 3 years, suggesting both early- and delayed-tolvaptan treated subjects benefitted to a similar degree.
A secondary endpoint testing the persistence of positive effects on renal function indicated that the preservation of eGFR observed by the end of the TEMPO 3:4 pivotal trial (3.01 to 3.34 mL/min/1.73 m2 at follow-up visits 1 and 2) could be preserved during open-label treatment This difference was maintained in the pre-specified MMRM analysis (3.15 mL/min/1.73 m2, 95% CI 1.462 to 4.836, p = 0.0003) and with sensitivity analyses where baseline eGFR data were carried forward (2.64 mL/min/1.73 m2, 95% CI 0.672 to 4.603, p = 0.0086). These data suggest that Tolvaptan (JINARC®) can slow the rate of renal function decline, and that these benefits persist over the duration of therapy.
Longer term data are not currently available to show whether long-term therapy with Tolvaptan (JINARC®) continues to slow the rate of renal function decline and affect clinical outcomes of ADPKD, including delay in the onset of end-stage renal disease.
Genotyping for PKD1 and PKD2 genes was conducted in a majority of patients entering the open-label extension study (TEMPO 4:4) but the results are not yet known.
Following an additional 2 years of tolvaptan treatment, resulting in a total of 5 years on tolvaptan therapy no new safety signals were identified.
The phase 3, multi-centre, international, randomized-withdrawal, placebo-controlled, double-blind trial 156-13-210 compared the efficacy and safety of tolvaptan (45 to 120 mg/day) to placebo in patients able to tolerate tolvaptan during a five-week titration and run-in period on tolvaptan. The trial utilized a randomized withdrawal design, to enrich for patients that were able to tolerate tolvaptan for a 5-week, single-blind prerandomization period consisting of a 2-week titration period and 3-week run-in period. The design was used to minimize the impact of early discontinuation and missing data on trial endpoints.
A total of 1,370 patients (age 18-65) with chronic kidney disease (CKD) with an eGFR between 25 and 65 mL/min/1.73 m2 if younger than age 56; or eGFR between 25 and 44 mL/min/1.73 m2, plus eGFR decline >2.0 mL/min/1.73 m2/year if between age 56-65 were randomized to either tolvaptan (n = 683) or placebo (n = 687) and were treated for a period of 12 months.
For subjects randomized, the baseline, average estimated glomerular filtration rate (eGFR) was 41 mL/min/1.73 m2 (CKD-Epidemiology formula) and historical TKV, available in 318 (23%) of subjects, averaged 2,026 mL. Approximately 5%, 75% and 20% had an eGFR 60 mL/min/1.73 m2 or greater (CKD stage 2), or less than 60 and greater than 30 mL/min/1.73 m2 (CKD stage 3) or less than 30 but greater than 15 mL/min/1.73 m2 (CKD stage 4), respectively. The CKD stage 3 can be subdivided further to stage 3a 30 %, (eGFR 45 mL/min/1.73 m2 to less than 60 mL/min/1.73 m2) and stage 3b 45%, (eGFR between 30-45 mL/min/1.73 m2).
The primary endpoint of the trial was the change in estimated glomerular filtration rate (eGFR) from pre-treatment baseline levels to post-treatment assessment. In patients treated with tolvaptan the reduction in eGFR was significantly less than in patients treated with placebo (p < 0.0001). The treatment difference in eGFR change observed in this trial is 1.27 mL/min/1.73 m2, representing a 35 % reduction in the LS means of change in eGFR of -2.34 mL/min/1.73 m2 in tolvaptan group relative to a -3.61 mL/min/1.73 m2 in placebo group observed over the course of one year. The key secondary endpoint was a comparison of the efficacy of tolvaptan treatment vs. placebo in reducing the decline of annualized eGFR slope across all measured time points in the trial. These data also showed significant benefit from tolvaptan vs. placebo (p <0.0001).
Subgroup analysis of the primary and secondary endpoints by CKD stage found similar, consistent treatment effects relative to placebo for subjects in stages 2, 3a, 3b and 4 at baseline.
A pre-specified subgroup analysis suggested that tolvaptan had less of an effect in patients older than 55 years of age, a small subgroup with a notably slower rate of eGFR decline.
Paediatric population: The European Medicines Agency has deferred the obligation to submit the results of studies with tolvaptan in one or more subsets of the paediatric population in polycystic kidney disease (see Dosage & Administration for information on paediatric use).
Pharmacokinetics: Absorption: After oral administration, tolvaptan is rapidly absorbed with peak plasma concentrations occurring about 2 hours after dosing. The absolute bioavailability of tolvaptan is about 56 %. Co-administration of tolvaptan with a high-fat meal increased peak concentrations of tolvaptan up to 2-fold but left AUC unchanged. Even though the clinical relevance of this finding is not known, to minimise the unnecessary risk of increasing the maximal exposure the morning dose should be taken under fasted conditions (see Dosage & Administration).
Distribution: Following single oral doses of ≥ 300 mg, peak plasma concentrations appear to plateau, possibly due to saturation of absorption. Tolvaptan binds reversibly (98 %) to plasma proteins.
Biotransformation: Tolvaptan is extensively metabolised in the liver almost exclusively by CYP3A. Tolvaptan is a weak CYP3A4 substrate and does not appear to have any inhibitory activity. In vitro studies indicated that tolvaptan has no inhibitory activity for CYP3A. Fourteen metabolites have been identified in plasma, urine and faeces; all but one were also metabolised by CYP3A. Only the oxobutyric acid metabolite is present at greater than 10 % of total plasma radioactivity; all others are present at lower concentrations than tolvaptan. Tolvaptan metabolites have little to no contribution to the pharmacological effect of tolvaptan; all metabolites have no or weak antagonist activity for human V2 receptors when compared with tolvaptan. The terminal elimination half-life is about 8 hours and steady-state concentrations of tolvaptan are obtained after the first dose.
Elimination: Less than 1 % of intact active substance is excreted unchanged in the urine. Radio labelled tolvaptan experiments showed that 40% of the radioactivity was recovered in the urine and 59 % was recovered in the faeces, where unchanged tolvaptan accounted for 32 % of radioactivity. Tolvaptan is only a minor component in plasma (3%).
Linearity: Following single oral doses, Cmax values show less than dose proportional increases from 30 to 240 mg and then a plateau at doses from 240 to 480 mg, AUC increases linearly.
Following multiple once daily dosing of 300 mg, tolvaptan exposure was only increased 6.4-fold when compared to a 30 mg dose. For split-dose regimens of 30, 60 and 120 mg/day in ADPKD patients, tolvaptan exposure (AUC) increases linearly.
Pharmacokinetics in special populations: Age: Clearance of tolvaptan is not significantly affected by age.
Hepatic impairment: The effect of mildly or moderately impaired hepatic function (Child-Pugh classes A and B) on the pharmacokinetics of tolvaptan was investigated in 87 patients with liver disease of various origins. No clinically significant changes have been seen in clearance for doses ranging from 5 to 60 mg. Very limited information is available in patients with severe hepatic impairment (Child-Pugh class C).
In a population pharmacokinetic analysis in patients with hepatic oedema, AUC of tolvaptan in severely (Child-Pugh class C) and mildly or moderately (Child-Pugh classes A and B) hepatic impaired patients were 3.1 and 2.3 times higher than that in healthy subjects.
Renal impairment: In a population pharmacokinetic analysis for patients with ADPKD, tolvaptan concentrations were increased, compared to healthy subjects, as renal function decreased below eGFR of 60 mL/min/1.73 m2. An eGFRCKD-EPI decrease from 72.2 to 9. 79 (mL/min/1.73 m2) was associated with a 32 % reduction in total body clearance.
Toxicology: Preclinical safety data: Non-clinical data revealed no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity or carcinogenic potential. Teratogenicity was noted in rabbits given 1,000 mg/kg/day (7.5 times the exposure from the 120 mg/day human dose on an AUC basis). No teratogenic effects were seen in rabbits at 300 mg/kg/day (about 1.25 to 2.65 times the exposure in humans at the 120 mg/day dose, based on AUC). In a peri- and post-natal study in rats, delayed ossification and reduced pup body weight were seen at the high dose of 1,000 mg/kg/day.
Two fertility studies in rats showed effects on the parental generation (decreased food consumption and body weight gain, salivation), but tolvaptan did not affect reproductive performance in males and there were no effects on the foetuses. In females, abnormal oestrus cycles were seen in both studies.
The no observed adverse effect level (NOAEL) for effects on reproduction in females (100 mg/kg/day) was about 8-times the maximum human recommended dose of 120 mg/day on a mg/m2 basis.
Tolvaptan (JINARC®) is indicated to slow the progression of cyst development and renal insufficiency of autosomal dominant polycystic kidney disease (ADPKD) in adults with CKD stage 1 to 4 at initiation of treatment with evidence of rapidly progressing disease (see Pharmacology: Pharmacodynamics under Actions).
Tolvaptan (JINARC
®) is to be administered twice daily in split dose regimens of 45 mg + 15 mg, 60 mg + 30 mg or 90 mg + 30 mg. The morning dose is to be taken at least 30 minutes before the morning meal. The second daily dose can be taken with or without food. According to these split dose regimens the total daily doses are 60, 90, or 120 mg.
Tablets must be swallowed without chewing and with a glass of water.
Dose titration: The initial dose is 60 mg tolvaptan per day as a split-dose regimen of 45 mg + 15 mg (45 mg taken upon waking and prior the morning meal and 15 mg taken 8 hours later). The initial dose is to be titrated upward to a split-dose regimen of 90 mg tolvaptan (60 mg + 30 mg) per day and then to a target split-dose regimen of 120 mg tolvaptan (90 mg + 30 mg) per day, if tolerated, with at least weekly intervals between titrations. Dose titration has to be performed cautiously to ensure that high doses are not poorly tolerated through overly rapid up-titration. Patients may down-titrate to lower doses based on tolerability. Patients have to be maintained on the highest tolerable tolvaptan dose.
The aim of dose titration is to block activity of vasopressin at the renal V2 receptor as completely and constantly as possible, while maintaining acceptable fluid balance (see Precautions).
Measurements of urine osmolality are recommended to monitor the adequacy of vasopressin inhibition. Periodic monitoring of plasma osmolality or serum sodium (to calculate plasma osmolarity) and/or body weight should be considered to monitor the risk of dehydration secondary to the aquaretic effects of tolvaptan in case of patient's insufficient water intake. The safety and efficacy of Tolvaptan (JINARC
®) in CKD stage 5 have not been adequately explored and therefore tolvaptan treatment should be discontinued if renal insufficiency progresses to CKD stage 5. The morning dose of Tolvaptan (JINARC
®) is to be taken at least 30 minutes before the morning meal. The second daily dose can be taken with or without food. Therapy must be interrupted if the ability to drink or the accessibility to water is limited (see Precautions).
PRECAUTION: Tolvaptan treatment must be initiated and monitored under the supervision of physicians with expertise in managing ADPKD and a full understanding of the risks of tolvaptan therapy including hepatic toxicity and monitoring requirements (see Precautions).
Tolvaptan must not be taken with grapefruit juice (see Interactions). Patients must be instructed to drink sufficient amounts of water or other aqueous fluids (see Precautions)
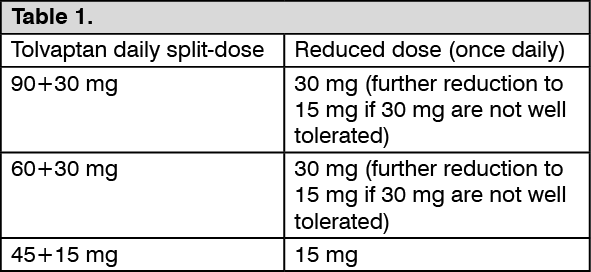
IMPORTANT PRECAUTION: Dose adjustment for patients taking strong CYP3A inhibitors: In patients taking strong CYP3A inhibitors (see Interactions), tolvaptan doses have to be reduced as follows: See Table 1.
 Click on icon to see table/diagram/image
Dose adjustment for patients taking moderate CYP3A inhibitors:
Click on icon to see table/diagram/image
Dose adjustment for patients taking moderate CYP3A inhibitors: In patients taking moderate CYP3A inhibitors, tolvaptan doses have to be reduced as follows: See Table 2.
Click on icon to see table/diagram/image
Further reductions have to be considered if patients cannot tolerate the reduced tolvaptan doses.
Elderly population: Increasing age has no effect on tolvaptan plasma concentrations. However, the safety and effectiveness of tolvaptan in ADPKD patients aged over 50 years has not yet been established.
Renal impairment: Tolvaptan is contraindicated in anuric patients (see Contraindications).
Dose adjustment is not required in patients with renal impairment. No clinical trials in subjects with a creatinine clearance < 10 mL/min or in patients undergoing dialysis have been conducted. The risk of hepatic damage in patients with severely reduced renal function (i.e. eGFR < 20) may be increased; these patients should be carefully monitored for hepatic toxicity. Data for patients in CKD stage 3 are more limited than for patients in stage 1 or 2 (see Pharmacology: Pharmacodynamics under Actions).
Hepatic impairment: In patients with severe hepatic impairment the benefits and risks of treatment with Tolvaptan (JINARC
®) must be evaluated carefully. Patients must be managed carefully and liver enzymes must be monitored regularly (see Precautions). Tolvaptan (JINARC
®) is contraindicated in patients with elevated liver enzymes and/or signs or symptoms of liver injury prior to initiation of treatment that meet the requirements for permanent discontinuation of tolvaptan (see Contraindications and Precautions). No dose adjustment is needed in patients with mild or moderate hepatic impairment (Child-Pugh classes A and B).
Paediatric population: The safety and efficacy of tolvaptan in children and adolescents has not yet been established. No data are available. Tolvaptan is not recommended in the paediatric age group.
Single oral doses up to 480 mg (4 times the maximum recommended daily dose) and multiple doses up to 300 mg once daily for 5 days have been well tolerated in trials in healthy subjects. There is no specific antidote for tolvaptan intoxication. The signs and symptoms of an acute overdose can be anticipated to be those of excessive pharmacologic effect: a rise in serum sodium concentration, polyuria, thirst and dehydration/hypovolemia.
No mortality was observed in rats or dogs following single oral doses of 2,000 mg/kg (maximum feasible dose). A single oral dose of 2,000 mg/kg was lethal in mice and symptoms of toxicity in affected mice included decreased locomotor activity, staggering gait, tremor and hypothermia.
In patients with suspected tolvaptan overdose, assessment of vital signs, electrolyte concentrations, ECG and fluid status is recommended. Appropriate replacement of water and/or electrolytes must continue until aquaresis abates. Dialysis may not be effective in removing tolvaptan because of its high binding affinity for human plasma protein (> 98 %).
Hypersensitivity to the active substance or to any of the excipients listed in Description or to benzazepine or benzazepine derivatives (see Precautions).
Elevated liver enzymes and/or signs or symptoms of liver injury prior to initiation of treatment that meet the requirements for permanent discontinuation of tolvaptan (see Precautions).
Anuria.
Volume depletion.
Hypernatraemia.
Patients who cannot perceive or respond to thirst.
Pregnancy (see Use in Pregnancy & Lactation).
Breast-feeding (see Use in Pregnancy & Lactation).
Idiosyncratic Hepatic Toxicity: Tolvaptan has been associated with idiosyncratic elevations of blood alanine and aspartate aminotransferases (ALT and AST) with infrequent cases of concomitant elevations in bilirubin-total (BT).
In post-marketing experience with tolvaptan in ADPKD, acute liver failure requiring liver transplantation has been reported.
In a double-blind, placebo-controlled trial in patients with ADPKD, elevation (> 3 x upper limit of normal [ULN]) of ALT was observed in 4.4 % (42/958) of patients on tolvaptan and 1.0 % (5/484) of patients on placebo, while elevation (> 3 x ULN) of AST was observed in 3.1 % (30/958) of patients on tolvaptan and 0.8 % (4/484) patients on placebo. Two (2/957, 0.2 %) of these tolvaptan treated-patients, as well as a third patient from an extension open label trial, exhibited increases in hepatic enzymes (> 3 x ULN) with concomitant elevations in BT (> 2 x ULN). The period of onset of hepatocellular injury (by ALT elevations > 3 x ULN) was within 3 to 14 months after initiating treatment and these increases were reversible, with ALT returning to < 3 x ULN within 1 to 4 months. While these concomitant elevations were reversible with prompt discontinuation of tolvaptan, they represent a potential for significant liver injury. Similar changes with other medicinal products have been associated with the potential to cause irreversible and potentially life-threatening liver injury.
Prescribing physicians must comply fully with the safety measures required as follows.
To mitigate the risk of significant and/or irreversible liver injury, blood testing for hepatic transaminases and bilirubin is required prior to initiation of Tolvaptan (JINARC®), continuing monthly for 18 months and at regular 3-monthly intervals thereafter. Concurrent monitoring for symptoms that may indicate liver injury (such as fatigue, anorexia, nausea, right upper abdominal discomfort, vomiting, fever, rash, pruritus, dark. urine or jaundice) is recommended.
If a patient shows abnormal ALT, AST or BT levels prior to initiation of treatment which fulfil the criteria for permanent discontinuation (see as follows) the use of tolvaptan is contraindicated (see Contraindications). In case of abnormal baseline levels below the limits for permanent discontinuation treatment can only be initiated if the potential benefits of treatment outweigh the potential risks and liver function testing must continue at increased time frequency. The advice of a hepatologist is recommended.
During the first 18 months of treatment, Tolvaptan (JINARC®) can only be supplied to patients whose physician has determined that liver function supports continued therapy.
At the onset of symptoms or signs consistent with hepatic injury or if clinically significant abnormal ALT or AST increases are detected during treatment, Tolvaptan (JINARC®) administration must be immediately interrupted and repeat tests including ALT, AST, BT and alkaline phosphatase (AP) must be obtained as soon as possible (ideally within 48-72 hours). Testing must continue at increased time frequency until symptoms/signs/laboratory abnormalities stabilise or resolve, at which point Tolvaptan (JINARC®) may be reinitiated.
Current clinical practice suggests that Tolvaptan (JINARC®) therapy is to be interrupted upon confirmation of sustained or increasing transaminase levels and permanently discontinued if significant increases and/or clinical symptoms of hepatic injury persist.
Recommended guidelines for permanent discontinuation include: ALT or AST > 8-times ULN; ALT or AST > 5-times ULN for more than 2 weeks; ALT or AST > 3-times ULN and (BT > 2-times ULN or International Normalized Ratio [INR] > 1.5); ALT or AST > 3-times ULN with persistent symptoms of hepatic injury as previously noted.
If ALT and AST levels remain below 3-times the upper limit of normal (ULN), Tolvaptan (JINARC®) therapy may be cautiously re-started, with frequent monitoring at the same or lower doses, as transaminase levels appear to stabilise during continued therapy in some patients.
Access to water: Tolvaptan may cause adverse reactions related to water loss such as thirst, polyuria, nocturia, and pollakiuria (see Adverse Reactions). Therefore, patients must have access to water (or other aqueous fluids) and be able to drink sufficient amounts of these fluids (see Dosage &Administration). Patients have to be instructed to drink water or other aqueous fluids at the first sign of thirst in order to avoid excessive thirst or dehydration.
Additionally, patients have to drink 1-2 glasses of fluid before bedtime regardless of perceived thirst and replenish fluids overnight with each episode of nocturia.
Dehydration: Volume status must be monitored in patients taking tolvaptan because treatment with tolvaptan may result in severe dehydration which constitutes a risk factor for renal dysfunction. If dehydration becomes evident, take appropriate action which may include the need to interrupt or reduce the dose of tolvaptan and increase fluid intake. Special care must be taken in patients having diseases that impair appropriate fluid intake or who are at an increased risk of water loss e.g. in case of vomiting or diarrhoea.
Urinary outflow obstruction: Urinary output must be secured. Patients with partial obstruction of urinary outflow, for example patients with prostatic hypertrophy or impairment of micturition, have an increased risk of developing acute retention.
Fluid and electrolyte balance: Fluid and electrolyte status must be monitored in all patients. Administration of tolvaptan induces copious aquaresis and may cause dehydration and increases in serum sodium (see Adverse Reactions) and is contraindicated in hypernatraemic patients (see Contraindications). Therefore, serum creatinine, electrolytes and symptoms of electrolyte imbalances (e.g. dizziness, fainting, palpitations, confusion, weakness, gait instability, hyper-reflexia, seizures, coma) have to be assessed prior to and after starting tolvaptan to monitor for dehydration.
During long-term treatment electrolytes have to be monitored at least every three months.
Serum sodium abnormalities: Pre-treatment sodium abnormalities (hyponatraemia or hypernatraemia) must be corrected prior to initiation with tolvaptan therapy.
Anaphylaxis: In post-marketing experience, anaphylaxis (including anaphylactic shock and rash generalised) has been reported very rarely following administration of tolvaptan. This type of reaction occurred after the first administration of tolvaptan. Patients have to be carefully monitored during treatment. Patients with known hypersensitivity reactions to benzazepines or benzazepine derivatives (e.g. benazepril, conivaptan, fenoldopam mesylate or mirtazapine) may be at risk for hypersensitivity reaction to tolvaptan (see Contraindications).
If an anaphylactic reaction or other serious allergic reactions occur, administration of tolvaptan must be discontinued immediately and appropriate therapy initiated. Since hypersensitivity is a contraindication (see Contraindications) treatment must never be restarted after an anaphylactic reaction or other serious allergic reactions.
Lactose: Tolvaptan (JINARC®) contains lactose as an excipient. Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Diabetes mellitus: Diabetic patients with an elevated glucose concentration (e.g. in excess of 300 mg/dl) may present with pseudohyponatraemia. This condition must be excluded prior and during treatment with tolvaptan. Tolvaptan may cause hyperglycaemia (see Adverse Reactions). Therefore, diabetic patients treated with tolvaptan must be managed cautiously. In particular this applies to patients with inadequately controlled type II diabetes.
Uric acid increases: Decreased uric acid clearance by the kidney is a known effect of tolvaptan. In a double-blind, placebo-controlled trial of patients with ADPKD, potentially clinically significant increased uric acid (greater than 10 mg/dL) was reported at a higher rate in tolvaptan-patients (6.2%) compared to placebo-treated patients (1.7%). Adverse reactions of gout were reported more frequently in tolvaptan-treated patients (28/961, 2.9%) than in patients receiving placebo (7/483, 1.4%). In addition, increased use of allopurinol and other medicinal products used to manage gout were observed in the double-blind, placebo-controlled trial. Effects on serum uric acid are attributable to the reversible renal hemodynamic changes that occur in response to tolvaptan effects on urine osmolality and may be clinically relevant. However, events of increased uric acid and/or gout were not serious and did not cause discontinuation of therapy in the double-blind, placebo-controlled trial. Uric acid concentrations are to be evaluated prior to initiation of Tolvaptan (JINARC®) therapy, and as indicated during treatment based on symptoms.
Effect of tolvaptan on glomerular filtration rate (GFR): A reversible reduction in GFR has been observed in ADPKD trials at the initiation of tolvaptan treatment.
Effects on ability to drive and use machines: Tolvaptan (JINARC®) has minor influence on the ability to drive or use machines. However, when driving vehicles or using machines it has to be taken into account that occasionally dizziness, asthenia or fatigue may occur.
Pregnancy: There are no adequate data from the use of tolvaptan in pregnant women. Studies in animals have shown reproductive toxicity (see Pharmacology: Toxicology: Preclinical safety data under Actions). The potential risk for humans is unknown.
Women of childbearing potential must use adequate contraceptive measures during Tolvaptan (JINARC®) use. Tolvaptan (JINARC®) must not be used during pregnancy (see Contraindications).
Breast-feeding: It is unknown whether tolvaptan is excreted in human breast milk. Studies in rats have shown excretion of tolvaptan in milk.
The potential risk for humans is unknown. Tolvaptan (JINARC®) is contraindicated during breast-feeding (see Contraindications).
Fertility: Studies in animals showed effects on fertility (see Pharmacology: Toxicology: Preclinical safety data under Actions). The potential risk for humans is unknown.
The pharmacodynamically predictable and most commonly reported adverse reactions are thirst, polyuria, nocturia, and pollakiuria occurring in approximately 55%, 38%, 29% and 23% of patients, respectively. Furthermore, tolvaptan has been associated with idiosyncratic elevations of blood alanine and aspartate aminotransferases (ALT and AST) with infrequent cases of concomitant elevations in bilirubin-total (BT).
The incidences of the Adverse Drug Reactions (ADRs) associated with tolvaptan therapy are tabulated as follows. The table is based on adverse events reported during clinical trials and/or post-marketing use. All ADRs are listed by system organ class and frequency; very common (≥1/10), common (≥1/100 to <l/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare (<1/10,000) and not known (cannot be estimated from the available data). Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
The frequency of adverse reactions reported during post-marketing use cannot be determined as they are derived from spontaneous reports. Consequently, the frequency of these adverse events is qualified as "not known". (See Table 3.)
Click on icon to see table/diagram/image
To mitigate the risk of significant or irreversible liver injury, blood testing for hepatic transaminases is required prior to initiation of Tolvaptan (JINARC
®) treatment, continuing monthly for 18 months and at regular 3-monthly intervals thereafter (see Precautions).
The most frequent adverse reactions are related to water loss. It is therefore of greatest importance that patients have access to water and are able to drink sufficient amounts of fluids. The volume status of patients taking tolvaptan must be monitored to prevent dehydration (see Precautions).
Effect of other medicinal products on the pharmacokinetics of Tolvaptan: CYP3A inhibitors: Concomitant use of medicinal products that are moderate CYP3A inhibitors (e.g. amprenavir, aprepitant, atazanavir, ciprofloxacin, crizotinib, darunavir/ritonavir, diltiazem, erythromycin, fluconazole, fosamprenavir, imatinib, verapamil) or strong CYP3A inhibitors (e.g., itraconazole, ketoconazole, ritonavir, clarithromycin) increase tolvaptan exposure.
Co-administration of tolvaptan and ketoconazole resulted in a 440 % increase in area under time-concentration curve (AUC) and 248 % increase in maximum observed plasma concentration (Cmax) for tolvaptan.
Co-administration of tolvaptan and fluconazole, a moderate CYP3A inhibitor, produced a 200% and 80% increase in tolvaptan AUC and Cmax, respectively.
Co-administration of tolvaptan with grapefruit juice, a moderate to strong CYP3A inhibitor, produced a doubling of peak tolvaptan concentrations (Cmax).
Dose reduction of tolvaptan is recommended for patients while taking moderate or strong CYP3A inhibitors (see Dosage & Administration). Patients taking moderate or strong CYP3A inhibitors must be managed cautiously, in particular if the inhibitors are taken more frequently than once a day.
CYP3A inducers: Concomitant use of medicinal products that are potent CYP3A inducers (e.g., rifampicin) will decrease tolvaptan exposure and efficacy. Co-administration of tolvaptan with rifampicin reduces Cmax and AUC for tolvaptan by about 85 %. Therefore, concomitant administration of tolvaptan with potent CYP3A inducers (e.g., rifampicin, rifabutin, rifapentin, phenytoin, carbamazepine, and St. John's Wort) is to be avoided.
Co-administration with medicinal products that increase serum sodium concentration: There is no experience from controlled clinical trials with concomitant use of tolvaptan and hypertonic sodium chloride solution, oral sodium formulations, and medicinal products that increase serum sodium concentration. Medicinal products with high sodium content such as effervescent analgesic preparations and certain sodium containing treatments for dyspepsia may also increase serum sodium concentration. Concomitant use of tolvaptan with medicinal products that increase serum sodium concentration may result in a higher risk for developing hypernatraemia (see Precautions) and is therefore not recommended.
Diuretics: Tolvaptan has not been extensively studied in ADPKD in combination with diuretics. While there does not appear to be a synergistic or additive effect of concomitant use of tolvaptan with loop and thiazide diuretics, each class of agent has the potential to lead to severe dehydration, which constitutes a risk factor for renal dysfunction. If dehydration or renal dysfunction becomes evident, appropriate action must be taken which may include the need to interrupt or reduce doses of tolvaptan and/or diuretics and increased fluid intake. Other potential causes of renal dysfunction or dehydration must be evaluated and addressed.
Effect of tolvaptan on the pharmacokinetic of other products: CYP3A substrates: In healthy subjects, tolvaptan, a CYP3A substrate, had no effect on the plasma concentrations of some other CYP3A substrates (e.g. warfarin or amiodarone). Tolvaptan increased plasma levels of lovastatin by 1.3- to l.5-fold. Even though this increase has no clinical relevance, it indicates tolvaptan can potentially increase exposure to CYP3A4 substrates.
Transporter substrates: In-vitro studies indicate that tolvaptan is a substrate and competitive inhibitor of P-glycoprotein (P-gp). In-vitro studies indicate that tolvaptan or its oxobutyric metabolite may have the potential to inhibit OATP1B1, OATP1B3, OAT3, BCRP and OCT1 transporters. Steady state digoxin concentrations were increased (1.3-fold in maximum observed plasma concentration [Cmax] and 1.2-fold in area under the plasma concentration-time curve over the dosing interval [AUCτ]) when co-administered with multiple once daily 60 mg doses of tolvaptan. Patients receiving digoxin or other narrow therapeutic P-gp substrates (e.g., dabigatran) must therefore be managed cautiously and evaluated for excessive effects when treated with tolvaptan. Statins commonly used in the tolvaptan phase 3 pivotal trial (e.g., rosuvastatin and pitavastatin) are OATP1B1 or OATP1B3 substrates, however no difference in AE profile was observed during the phase 3 pivotal trial for tolvaptan in ADPKD. If OATP1B1 and OATP1B3 substrates (e.g., statins such as rosuvastatin and pitavastatin), OAT3 substrates (e.g., methotrexate, ciprofloxacin), BCRP substrates (e.g. sulfasalazine) or OCT1 substrates (e.g. metformin) are co-administered with tolvaptan, patients must be managed cautiously and evaluated for excessive effects of these medicinal products.
Diuretics or non-diuretic anti-hypertensive medicinal product(s): Standing blood pressure was not routinely measured in ADPKD trials, therefore a risk of orthostatic/postural hypotension due to a pharmacodynamic interaction with tolvaptan cannot be excluded.
Co-administration with vasopressin analogues: In addition to its renal aquaretic effect, tolvaptan is capable of blocking vascular vasopressin V2 receptors involved in the release of coagulation factors (e.g., von Willebrand factor) from endothelial cells. Therefore, the effect of vasopressin analogues such as desmopressin may be attenuated in patients using such analogues to prevent or control bleeding when co-administered with tolvaptan. It is not recommended to administer Tolvaptan (JINARC®) with vasopressin analogues.
Smoking and alcohol: Data related to smoking or alcohol history in ADPKD trials are too limited to determine possible interactions of smoking or alcohol with efficacy and safety of ADPKD treatment with tolvaptan.
Store at temperatures not exceeding 30°C.
C03XA01 - tolvaptan ; Belongs to the class of vasopressin antagonists. Used as diuretics.




 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image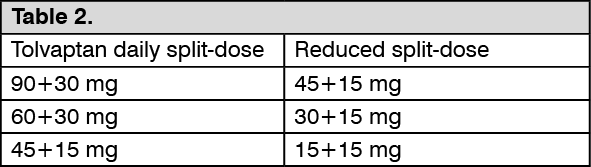 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image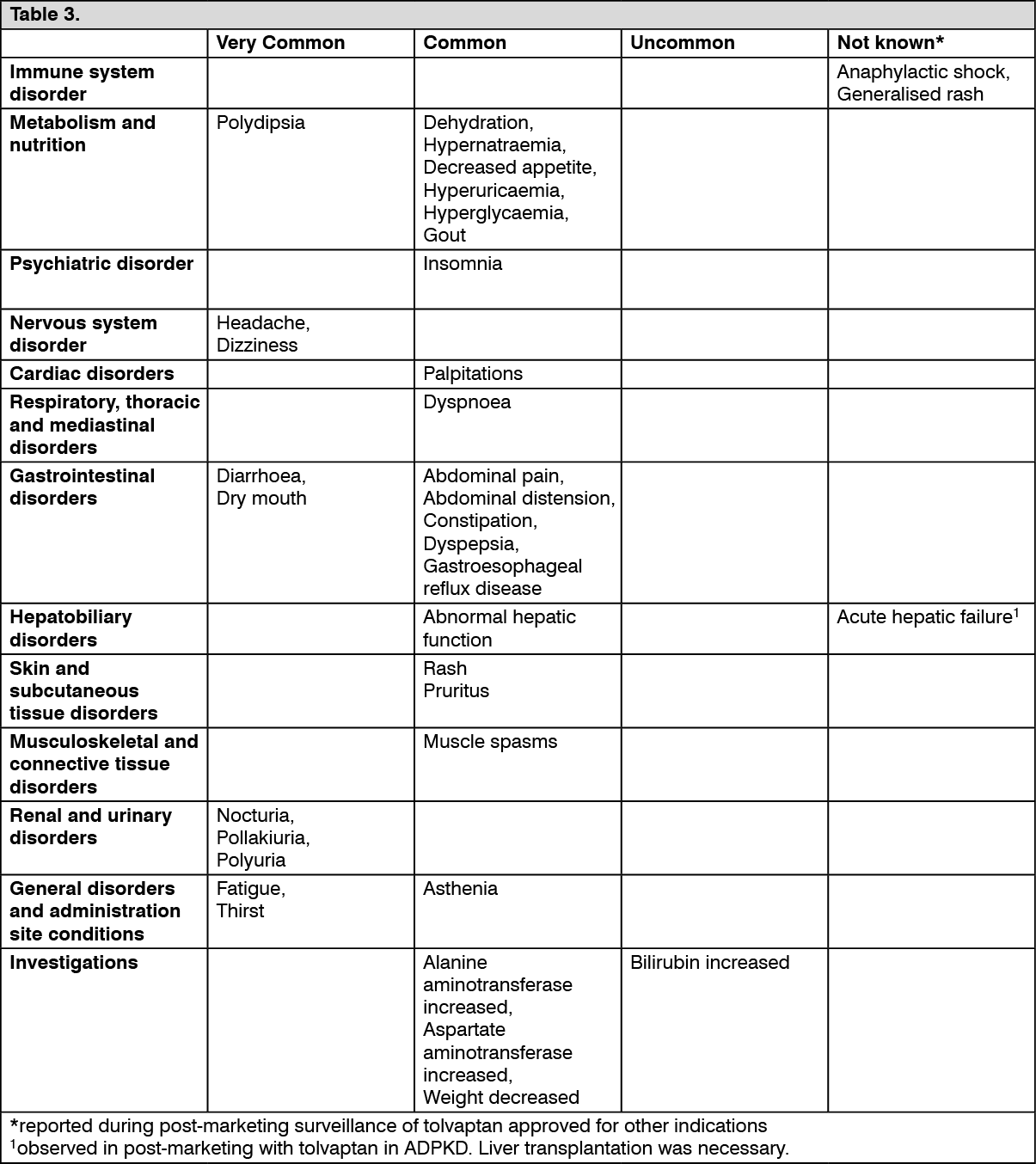 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
