


 Sign Out
Sign Out



Pharmacology: Pharmacodynamics: Mechanism of action: Nirmatrelvir is a peptidomimetic inhibitor of the SARS-CoV-2 main protease (Mpro), also referred to as 3C-like protease (3CLpro) or nsp5 protease. Inhibition of the SARS-CoV-2 Mpro renders the protein incapable of processing polyprotein precursors which leads to the prevention of viral replication.
Ritonavir inhibits the CYP3A-mediated metabolism of nirmatrelvir, thereby providing increased plasma concentrations of nirmatrelvir.
Antiviral activity: Nirmatrelvir exhibited antiviral activity against SARS-CoV-2 infection of differentiated normal human bronchial epithelial (dNHBE) cells, a primary human lung alveolar epithelial cell line (EC50 value of 61.8 nM and EC90 value of 181 nM) after 3 days of drug exposure. Nirmatrelvir had cell culture antiviral activity (with EC50 values in the low nanomolar range ≤3-fold relative to USA WA1/2020) against SARS CoV 2 isolates belonging to the Alpha (B.1.1.7), Gamma (P.1), Delta (B.1.617.2), Lambda (C.37), Mu (B.1.621) and Omicron (B.1.1.529/BA.1, BA.2, BA.2.12.1, BA.4, and BA.5) variants. The Beta (B.1.351) variant was the least susceptible tested variant with approximately 3.7-fold reduced susceptibility relative to the USA-WA1/2020 isolate.
Antiviral resistance in cell cultures and biochemical assays: SARS-CoV-2 Mpro residues potentially associated with nirmatrelvir resistance have been identified using a variety of methods, including SARS-CoV-2 resistance selection, testing of recombinant SARS-CoV-2 viruses with Mpro substitutions, and biochemical assays with recombinant SARS-CoV-2 Mpro containing amino acid substitutions. Table 1 indicates Mpro substitutions and combinations of Mpro substitutions that have been observed in nirmatrelvir selected SARS-CoV-2 in cell culture. Individual Mpro substitutions are listed regardless of whether they occurred alone or in combination with other Mpro substitutions. Note that the Mpro S301P and T304I substitutions overlap the P6 and P3 positions of the nsp5/nsp6 cleavage site located at the C-terminus of Mpro. Substitutions at other Mpro cleavage sites have not been associated with nirmatrelvir resistance in cell culture. The clinical significance of these substitutions is unknown. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageMost single Mpro mutations and some double mutations identified which reduced the susceptibility of SARS CoV 2 to nirmatrelvir resulted in an EC50 shift of <5-fold compared to wild type SARS CoV 2. In general, triple mutations and some double mutations led to EC50 changes of >5-fold to that of wild type. The clinical significance of these mutations needs to be further understood.
Viral load rebound and treatment-emergent mutations: Post-treatment viral nasal RNA rebounds were observed on Day 10 and/or Day 14 in a subset of Nirmatrelvir+Ritonavir (Paxlovid) and placebo recipients in EPIC-HR, irrespective of COVID-19 symptoms. The incidence of viral rebound in EPIC-HR occurred in both the Nirmatrelvir+Ritonavir (Paxlovid) treated participants and the untreated (placebo) participants, but at higher incidence in the Nirmatrelvir+Ritonavir (Paxlovid) arm (6.96% vs. 4.08%). So far, viral rebounds and symptoms recurrences of COVID-19 are not associated with more severe disease or emergence of resistance.
Clinical efficacy: The efficacy of Nirmatrelvir+Ritonavir (Paxlovid) is based on the interim analysis and the supporting final analysis of EPIC HR, a phase 2/3, randomized, double-blind, placebo-controlled study in non hospitalized, symptomatic adult participants with a laboratory confirmed diagnosis of SARS-CoV-2 infection. Eligible participants were 18 years of age and older with at least 1 of the following risk factors for progression to severe disease: diabetes, overweight (BMI >25), chronic lung disease (including asthma), chronic kidney disease, current smoker, immunosuppressive disease or immunosuppressive treatment, cardiovascular disease, hypertension, sickle cell disease, neurodevelopmental disorders, active cancer, medically related technological dependence, or were 60 years of age and older regardless of comorbidities. Participants with COVID-19 symptom onset of ≤5 days were included in the study. The study excluded individuals with a history of prior COVID-19 infection or vaccination.
Participants were randomized (1:1) to receive Nirmatrelvir+Ritonavir 300 mg + 100 mg (Paxlovid) or placebo orally every 12 hours for 5 days. The primary efficacy endpoint was the proportion of participants with COVID 19 related hospitalization or death from any cause through Day 28. The analysis was conducted in the modified intent-to-treat (mITT) analysis set (all treated participants with onset of symptoms ≤3 days who at baseline did not receive nor were expected to receive COVID-19 therapeutic mAb treatment), the mITT1 analysis set (all treated participants with onset of symptoms ≤5 days who at baseline did not receive nor were expected to receive COVID-19 therapeutic mAb treatment), and the mITT2 analysis set (all treated participants with onset of symptoms ≤5 days).
A total of 2246 participants were randomized to receive either Nirmatrelvir+Ritonavir (Paxlovid) or placebo. At baseline, mean age was 46 years with 13% of participants 65 years of age and older (3% were 75 years of age and older); 51% were male; 72% were White, 5% were Black or African American, and 14% were Asian; 45% were Hispanic or Latino; 66% of participants had onset of symptoms ≤3 days before initiation of study treatment; 81% had a BMI >25 kg/m2 (37% a BMI >30 kg/m2); 12% had diabetes mellitus; less than 1% of the study population had immune deficiency, 47% of participants were serological negative at baseline and 51% were serological positive. The mean (SD) baseline viral load was 4.63 log10 copies/mL (2.87); 26% of participants had a baseline viral load of >10^7 (copies/mL); 6.2% of participants either received or were expected to receive COVID-19 therapeutic mAb treatment at the time of randomization and were excluded from the mITT and mITT1 analyses. The primary SARS-CoV-2 variant across both treatment arms was Delta (98%), mostly clade 21J (based on interim analysis).
The baseline demographic and disease characteristics were balanced between the Nirmatrelvir+Ritonavir (Paxlovid) and placebo groups.
The determination of primary efficacy was based on a planned interim analysis of 774 participants in mITT population. The estimated risk reduction was -6.3% with unadjusted 95% CI of (-9.0%, -3.6%) and a 95% CI of (-10.61%, -2.02%) when adjusting for multiplicity. The 2-sided p-value was <0.0001 with 2-sided significance level of 0.002.
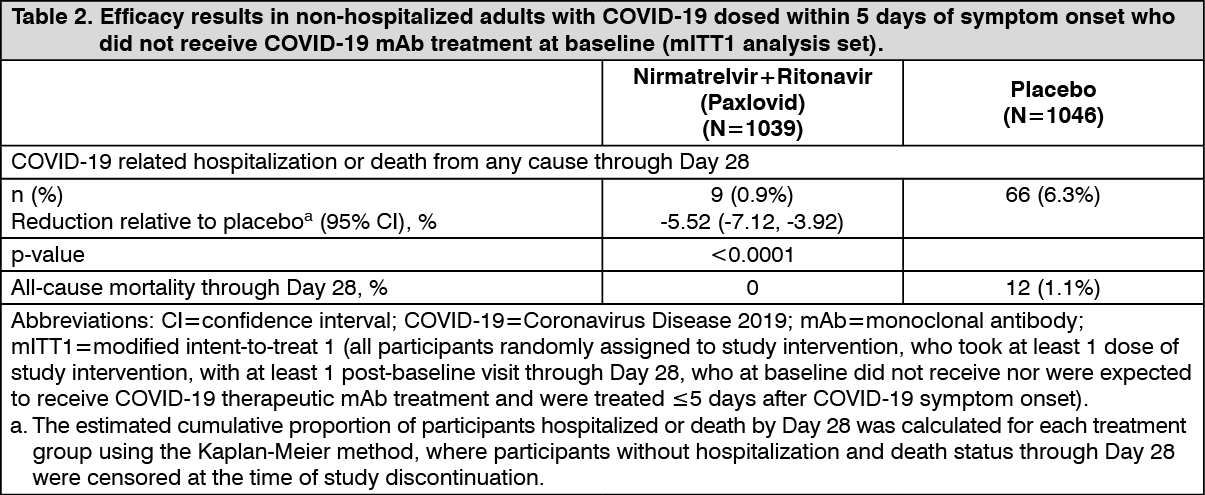
Table 2 provides results of the primary endpoint in the mITT1 analysis population for the full data set at final study completion. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe estimated risk reduction was -5.8% with 95% CI of (-7.8%, -3.8%) in participants dosed within 3 days of symptom onset, and -4.9% with 95% CI of (-7.7%, -2.2%) in the mITT1 subset of participants dosed >3 days from symptom onset.
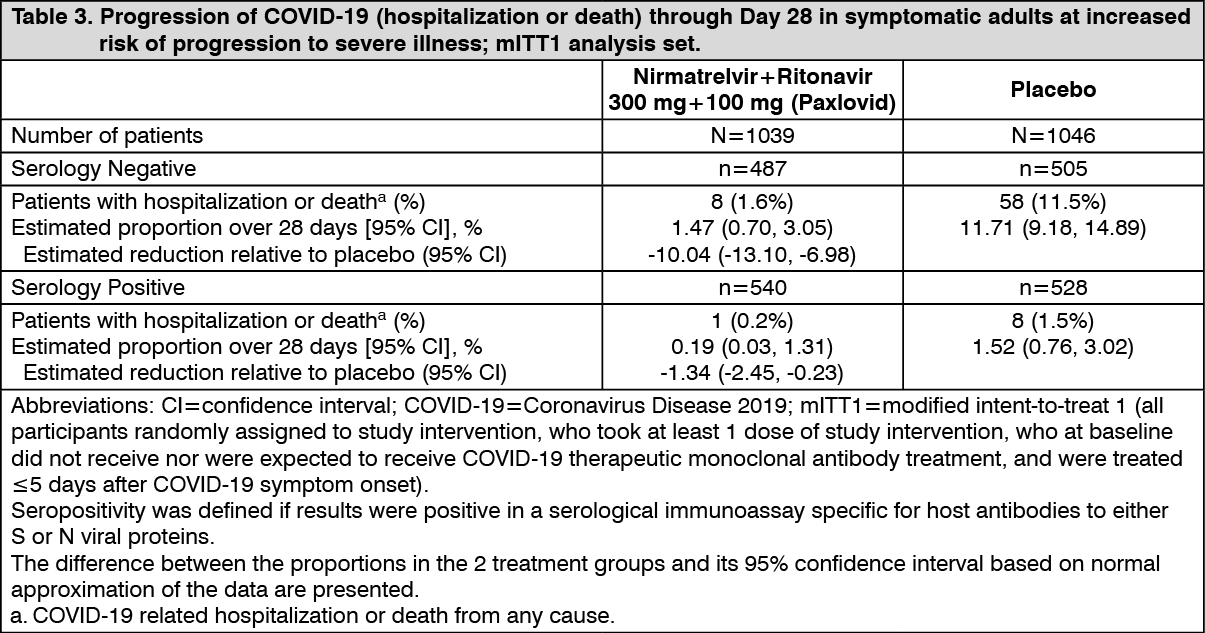
Consistent results were observed in the final mITT and mITT2 analysis populations. A total of 1379 participants were included in the mITT analysis population. The event rates were 5/697 (0.72%) in the Nirmatrelvir+Ritonavir (Paxlovid) group, and 44/682 (6.45%) in the placebo group. (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageEfficacy results for mITT1 were consistent across subgroups of participants including age (≥65 years) and BMI (BMI >25 and BMI >30) and diabetes.
Pediatric population: The European Medicines Agency has deferred the obligation to submit the results of studies with Nirmatrelvir+Ritonavir (Paxlovid) in one or more subsets of the pediatric population in treatment of COVID-19 (see Pediatric population under Dosage & Administration).
Pharmacokinetics: The pharmacokinetics of nirmatrelvir/ritonavir have been studied in healthy participants and in participants with mild-to-moderate COVID-19.
Ritonavir is administered with nirmatrelvir as a pharmacokinetic enhancer resulting in higher systemic concentrations and longer half-life of nirmatrelvir.
Upon repeat-dose of nirmatrelvir/ritonavir 75 mg/100 mg, 250 mg/100 mg, and 500 mg/100 mg administered twice daily, the increase in systemic exposure at steady-state appears to be less than dose proportional. Multiple dosing over 10 days achieved steady state on Day 2 with approximately 2-fold accumulation. Systemic exposures on Day 5 were similar to Day 10 across all doses.
Absorption: Following oral administration of nirmatrelvir/ritonavir 300 mg/100 mg after a single dose, the geometric mean nirmatrelvir Cmax and AUCinf at steady-state was 2.21 µg/mL and 23.01 µg*hr/mL, respectively. The median time to Cmax (Tmax) was 3.00 hrs. The arithmetic mean terminal elimination half-life was 6.1 hours.
Following oral administration of nirmatrelvir/ritonavir 300 mg/100 mg after a single dose, the geometric mean ritonavir Cmax and AUCinf was 0.36 µg/mL and 3.60 µg*hr/mL, respectively. The median time to Cmax (Tmax) was 3.98 hrs. The arithmetic mean terminal elimination half-life was 6.1 hours.
Effect of food on oral absorption: Dosing with a high fat meal increased the exposure of nirmatrelvir (approximately 61% increase in mean Cmax and 20% increase in mean AUClast) relative to fasting conditions following administration of 300 mg nirmatrelvir (2 × 150 mg)/100 mg ritonavir tablets.
Distribution: The protein binding of nirmatrelvir in human plasma is approximately 69%.
The protein binding of ritonavir in human plasma is approximately 98-99%.
Biotransformation: In vitro studies assessing nirmatrelvir without concomitant ritonavir suggest that nirmatrelvir is primarily metabolized by cytochrome P450 (CYP) 3A4. However, administration of nirmatrelvir with ritonavir inhibits the metabolism of nirmatrelvir. In plasma, the only medicinal product-related entity observed was unchanged nirmatrelvir. Minor oxidative metabolites were observed in the feces and urine.
In vitro studies utilizing human liver microsomes have demonstrated that CYP3A is the major isoform involved in ritonavir metabolism, although CYP2D6 also contributes to the formation of oxidation metabolite M-2.
Elimination: The primary route of elimination of nirmatrelvir when administered with ritonavir was renal excretion of intact medicinal product. Approximately 49.6% and 35.3% of the administered dose of nirmatrelvir 300 mg was recovered in urine and feces, respectively. Nirmatrelvir was the predominant drug related entity with small amounts of metabolites arising from hydrolysis reactions in excreta. In plasma, the only drug-related entity quantifiable was unchanged nirmatrelvir.
Human studies with radiolabelled ritonavir demonstrated that the elimination of ritonavir was primarily via the hepatobiliary system; approximately 86% of radiolabel was recovered from stool, part of which is expected to be unabsorbed ritonavir.
Specific populations: Age and gender: The pharmacokinetics of nirmatrelvir/ritonavir based on age and gender have not been evaluated.
Racial or ethnic groups: Systemic exposure in Japanese participants was numerically lower but not clinically meaningfully different than those in Western participants.
Patients with renal impairment: Compared to healthy controls with no renal impairment, the Cmax and AUC of nirmatrelvir in patients with mild renal impairment was 30% and 24% higher, in patients with moderate renal impairment was 38% and 87% higher, and in patients with severe renal impairment was 48% and 204% higher, respectively.
Patients with hepatic impairment: Compared to healthy controls with no hepatic impairment, the pharmacokinetics of nirmatrelvir in participants with moderate hepatic impairment was not significantly different. Adjusted geometric mean ratio (90% CI) of AUCinf and Cmax of nirmatrelvir comparing moderate hepatic impairment (test) to normal hepatic function (reference) was 98.78% (70.65%, 138.12%) and 101.96% (74.20%, 140.11%), respectively.
Nirmatrelvir/ritonavir has not been studied in patients with severe hepatic impairment.
Interaction studies conducted with nirmatrelvir/ritonavir: CYP3A4 was the major contributor to the oxidative metabolism of nirmatrelvir when nirmatrelvir was tested alone in human liver microsomes. Ritonavir is an inhibitor of CYP3A and increases plasma concentrations of nirmatrelvir and other drugs that are primarily metabolized by CYP3A. Despite being coadministered with ritonavir as a pharmacokinetic enhancer, there is potential for strong inhibitors and inducers to alter the pharmacokinetics of nirmatrelvir.
Nirmatrelvir does not reversibly inhibit CYP2D6, CYP2C9, CYP2C19, CYP2C8, or CYP1A2 in vitro at clinically relevant concentrations. In vitro study results showed nirmatrelvir may be inducer of CYP3A4, CYP2B6, CYP2C8 and CYP2C9. The clinical relevance is unknown. Based on in vitro data, nirmatrelvir has a low potential to inhibit BCRP, MATE2K, OAT1, OAT3, OATP1B3 and OCT2. There is a potential for nirmatrelvir to inhibit MDR1, MATE1, OCT1 and OATP1B1 at clinically relevant concentrations.
The effect on the pharmacokinetics of nirmatrelvir/ritonavir was assessed with itraconazole (CYP3A inhibitor) and carbamazepine (CYP3A inducer). The test/reference ratios of the adjusted geometric means for nirmatrelvir AUCinf and Cmax were 44.50% and 56.82%, respectively, following nirmatrelvir/ritonavir 300 mg/100 mg coadministration with multiple oral doses of carbamazepine. The test/reference ratios of the adjusted geometric means for nirmatrelvir AUCtau and Cmax were 138.82% and 118.57%, respectively, when nirmatrelvir/ritonavir was coadministered with multiple doses of itraconazole as compared to nirmatrelvir/ritonavir administered alone.
The effect of nirmatrelvir/ritonavir on other drugs was assessed with midazolam (CYP3A substrate) and dabigatran (P-gp substrate). The test/reference ratios of the adjusted geometric means for midazolam AUCinf and Cmax were 1430.02% and 368.33%, respectively, when midazolam was coadministered with multiple doses of nirmatrelvir/ritonavir compared to midazolam administered alone. The test/reference ratios of the adjusted geometric means for dabigatran AUCinf and Cmax were 194.47% and 233.06%, respectively, following dabigatran administration with multiple doses of nirmatrelvir/ritonavir as compared to administration of dabigatran alone.
Toxicology: Preclinical Safety Data: No nonclinical safety studies have been conducted with nirmatrelvir in combination with ritonavir.
Nirmatrelvir: Studies of repeated dose toxicity and genotoxicity revealed no risk due to nirmatrelvir. No adverse effects were observed in fertility, embryo-fetal development, or pre- and postnatal development studies in rats. A study in pregnant rabbits showed an adverse decrease in fetal body weight, in the absence of significant maternal toxicity. Systemic exposure (AUC24) in rabbits at the maximum dose without adverse effect in fetal body weight was estimated to be approximately 3 times higher than exposure in humans at recommended therapeutic dose of Nirmatrelvir+Ritonavir (Paxlovid).
No carcinogenicity studies have been conducted with nirmatrelvir.
Ritonavir: Repeat-dose toxicity studies of ritonavir in animals identified major target organs as the liver, retina, thyroid gland and kidney. Hepatic changes involved hepatocellular, biliary and phagocytic elements and were accompanied by increases in hepatic enzymes. Hyperplasia of the retinal pigment epithelium and retinal degeneration have been seen in all of the rodent studies conducted with ritonavir, but have not been seen in dogs. Ultrastructural evidence suggests that these retinal changes may be secondary to phospholipidosis. However, clinical trials revealed no evidence of medicinal product-induced ocular changes in humans. All thyroid changes were reversible upon discontinuation of ritonavir. Clinical investigation in humans has revealed no clinically significant alteration in thyroid function tests.
Renal changes including tubular degeneration, chronic inflammation and proteinuria were noted in rats and are considered to be attributable to species specific spontaneous disease. Furthermore, no clinically significant renal abnormalities were noted in clinical trials.
Genotoxicity studies revealed no risk due to ritonavir. Long-term carcinogenicity studies of ritonavir in mice and rats revealed tumorigenic potential specific for these species, but are regarded as of no relevance for humans. Ritonavir produced no effects on fertility in rats. Developmental toxicity observed in rats (embryo-lethality, decreased fetal body weight and ossification delays and visceral changes, including delayed testicular descent) occurred mainly at a maternally toxic dosage. Developmental toxicity in rabbits (embryo-lethality, decreased litter size and decreased fetal weights) occurred at a maternally toxic dosage.