


 Sign Out
Sign Out
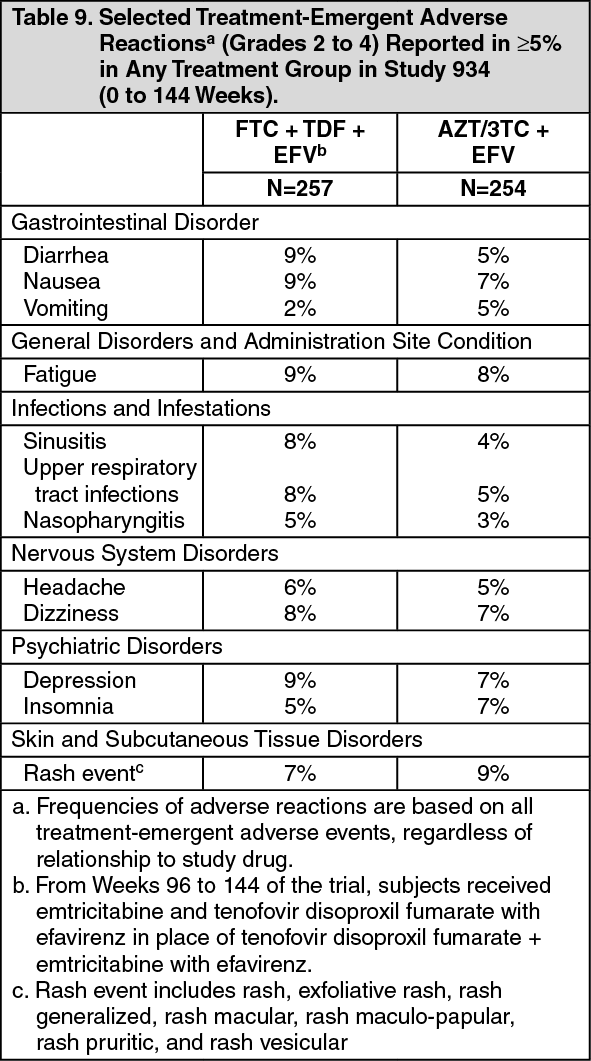
Clinical Trials in Adult Subjects: The most common adverse reactions (incidence greater than or equal to 10%, any severity) occurring in Study 934, an active-controlled clinical trial of efavirenz, emtricitabine, and tenofovir disoproxil fumarate, include diarrhea, nausea, fatigue, headache, dizziness, depression, insomnia, abnormal dreams, and rash. See also Table 9 for the frequency of treatment-emergent adverse reactions (Grades 2 to 4) occurring in greater than or equal to 5% of subjects treated in any treatment group in this trial.
Skin discoloration, manifested by hyperpigmentation on the palms and/or soles was generally mild and asymptomatic. The mechanism and clinical significance are unknown.
Study 934: Treatment Emergent Adverse Reactions: In Study 934, 511 antiretroviral-naive subjects received either tenofovir disoproxil fumarate + emtricitabine administered in combination with efavirenz (N=257) or zidovudine/lamivudine administered in combination with efavirenz (N=254) for 144 weeks. Subjects had a mean age of 40 years (range 20 to 73 years) and were predominantly male (88%). Overall, 65% were White, 17% were Black, and 13% were Hispanic. Adverse reactions observed in this trial were generally consistent with those seen in other trials in treatment-experienced or treatment-naive subjects receiving tenofovir disoproxil fumarate and/or emtricitabine (see Table 9).
 Click on icon to see table/diagram/image
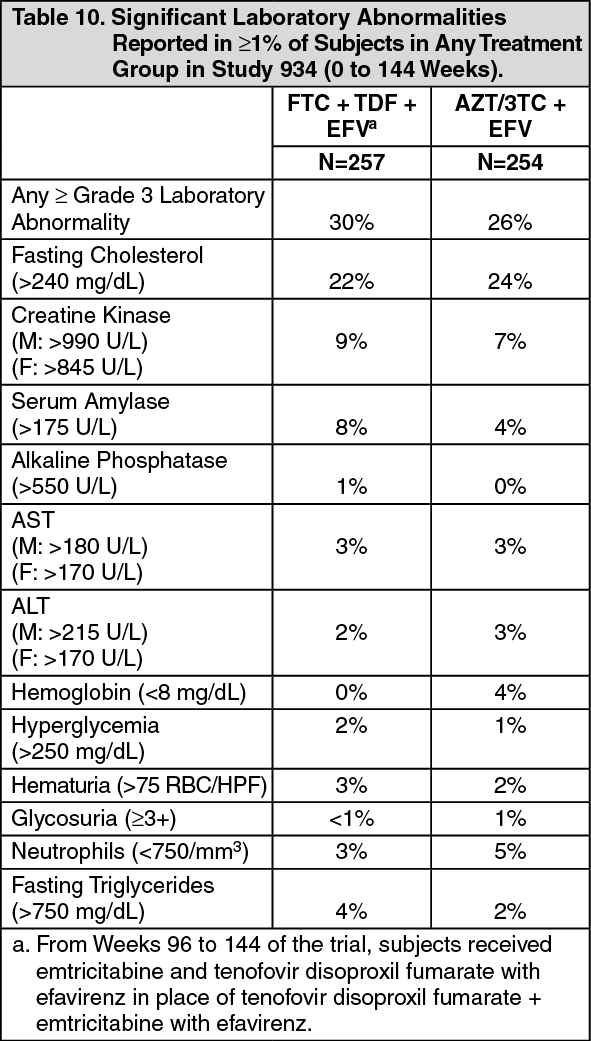
Click on icon to see table/diagram/imageLaboratory Abnormalities: Laboratory abnormalities observed in this trial were generally consistent with those seen in other trials of tenofovir disoproxil fumarate and/or emtricitabine (see Table 10).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn addition to the events described above for Study 934, other adverse reactions that occurred in at least 5% of subjects receiving emtricitabine or tenofovir disoproxil fumarate with other antiretroviral agents in clinical trials include anxiety, arthralgia, increased cough, dyspepsia, fever, myalgia, pain, abdominal pain, back pain, paresthesia, peripheral neuropathy (including peripheral neuritis and neuropathy), pneumonia, and rhinitis.
In addition to the laboratory abnormalities described above for Study 934, Grade 3 to 4 laboratory abnormalities of increased bilirubin (>2.5 x ULN), increased pancreatic amylase (>2 x ULN), increased or decreased serum glucose (<40 or >250 mg/dL), and increased serum lipase (>2 x ULN) occurred in up to 3% of subjects treated with emtricitabine or tenofovir disoproxil fumarate with other antiretroviral agents in clinical trials.
Clinical Trials in Pediatric Subjects: Emtricitabine: In addition to the adverse reactions reported in adults, anemia and hyperpigmentation were observed in 7% and 32%, respectively, of pediatric subjects (3 months to less than 18 years of age) who received treatment with emtricitabine in the larger of two open-label, uncontrolled pediatric trials (N=116). For additional information, please consult the emtricitabine prescribing information.
Tenofovir Disoproxil Fumarate: In pediatric clinical trials (Studies 352 and 321) conducted in 184 HIV-1 infected subjects 2 to less than 18 years of age, the adverse reactions observed in pediatric subjects who received treatment with tenofovir disoproxil fumarate were consistent with those observed in clinical trials of tenofovir disoproxil fumarate in adults.
Eighty-nine pediatric subjects (2 to less than 12 years of age) received tenofovir disoproxil fumarate in Study 352 for a median exposure of 104 weeks. Of these, 4 subjects discontinued from the trial due to adverse reactions consistent with proximal renal tubulopathy. Three of these 4 subjects presented with hypophosphatemia and also had decreases in total body or spine BMD Z score (see Precautions).
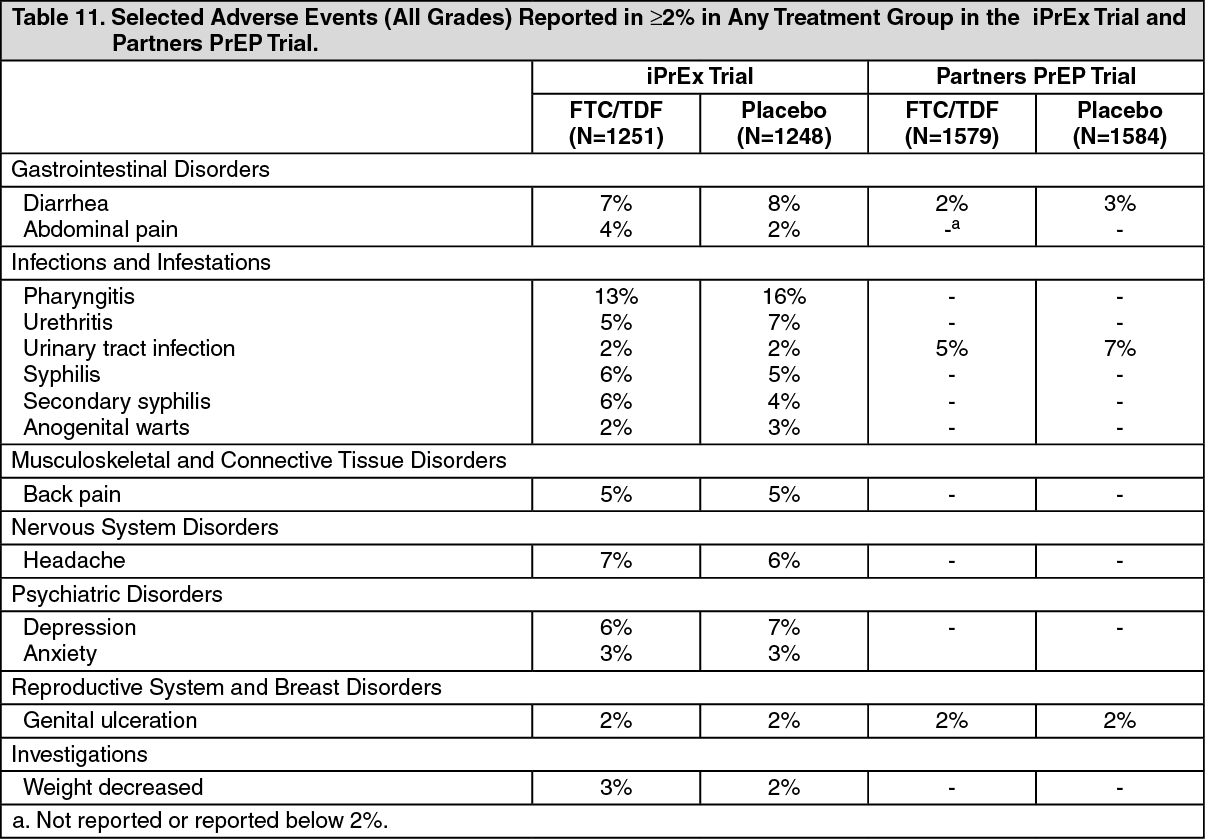
Adverse Reactions from Clinical Trial Experience in HIV-1 Uninfected Adult Subjects: No new adverse reactions to emtricitabine and tenofovir disoproxil fumarate were identified from two randomized placebo-controlled clinical trials (iPrEx, Partners PrEP) in which 2830 HIV-1 uninfected adults received emtricitabine and tenofovir disoproxil fumarate once daily for pre-exposure prophylaxis. Subjects were followed for a median of 71 weeks and 87 weeks, respectively. These trials enrolled HIV-negative individuals ranging in age from 18 to 67 years. The iPrEx trial enrolled only males or transgender females of Hispanic/Latino (72%), White (18%), Black (9%) and Asian (5%) race. The Partners PrEP trial enrolled both males (61 to 64% across treatment groups) and females in Kenya and Uganda. Table 11 provides a list of all adverse events that occurred ³2% of subjects in any treatment group in the iPrEx and Partners PrEP trials.
Laboratory Abnormalities: Table 12 provides a list of laboratory abnormalities observed in both trials. Six subjects in the TDF-containing arms of the Partners PrEP trial discontinued participation in the study due to an increase in blood creatinine compared with no discontinuations in the placebo group. One subject in the emtricitabine and tenofovir disoproxil fumarate arm of the iPrEx trial discontinued from the study due to an increase in blood creatinine and another due to low phosphorous.
In addition to the laboratory abnormalities described above, Grade 1 proteinuria (1+) occurred in 6% of subjects receiving emtricitabine and tenofovir disoproxil fumarate in the iPrEx trial. Grade 2 to 3 proteinuria (2 to 4+) and glycosuria (3+) occurred in less than 1% of subjects treated with emtricitabine and tenofovir disoproxil fumarate in the iPrEx trial and Partners PrEP trial. (See Tables 11 and 12).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image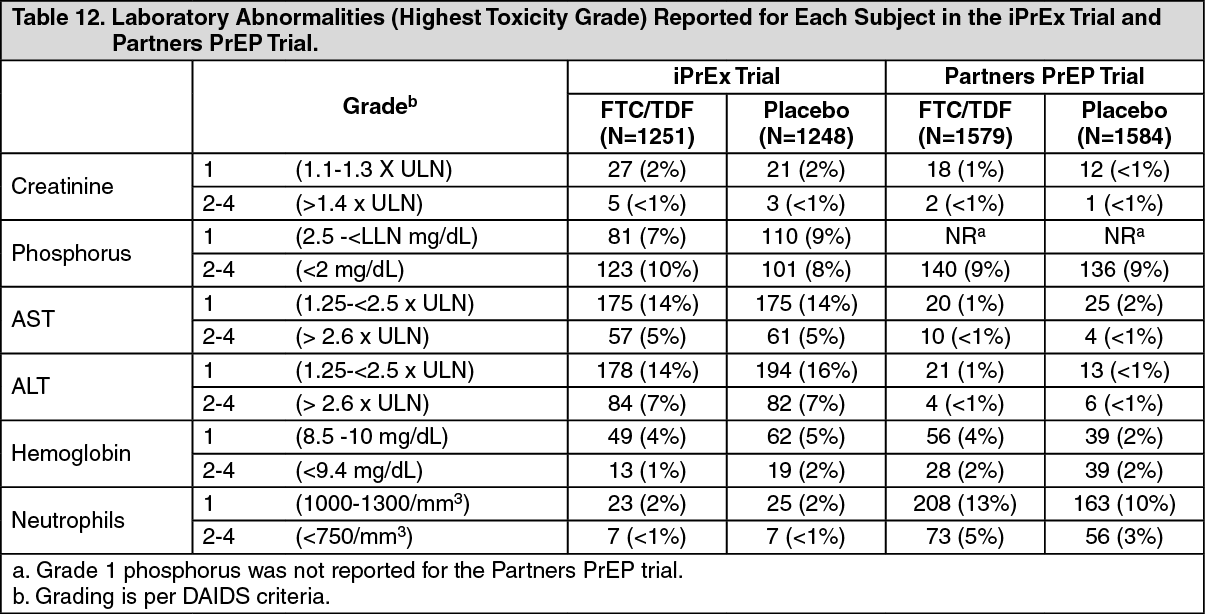 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageChanges in Bone Mineral Density: In clinical trials of HIV-1 uninfected individuals, decreases in BMD were observed. In the iPrEx trial, a substudy of 503 subjects found mean changes from baseline in BMD ranging from -0.4% to -1% across total hip, spine, femoral neck, and trochanter in the emtricitabine and tenofovir disoproxil fumarate group compared with the placebo group, which returned toward baseline after discontinuation of treatment. Thirteen percent of subjects receiving emtricitabine and tenofovir disoproxil fumarate vs. 6% of subjects receiving placebo lost at least 5% of BMD at the spine during treatment. Bone fractures were reported in 1.7% of the emtricitabine and tenofovir disoproxil fumarate group compared with 1.4% in the placebo group. No correlation between BMD and fractures was noted [See Pharmacology: Pharmacodynamics: Clinical Studies under Actions]. The Partners PrEP trial found similar fracture rates between treatment and placebo groups (0.8% and 0.6%, respectively). No BMD evaluations were conducted during this trial (see Pharmacology: Pharmacodynamics: Clinical Studies under Actions).
Postmarketing Experience: The following adverse reactions have been identified during postapproval use of tenofovir disoproxil fumarate. No additional adverse reactions have been identified during postapproval use of emtricitabine. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders: Allergic reaction, including angioedema.
Metabolism and Nutrition Disorders: Lactic acidosis, hypokalemia, hypophosphatemia.
Respiratory, Thoracic, and Mediastinal Disorders: Dyspnea.
Gastrointestinal Disorders: Pancreatitis, increased amylase, abdominal pain.
Hepatobiliary Disorders: Hepatic steatosis, hepatitis, increased liver enzymes (most commonly AST, ALT gamma GT).
Skin and Subcutaneous Tissue Disorders: Rash.
Musculoskeletal and Connective Tissue Disorders: Rhabdomyolysis, osteomalacia (manifested as bone pain and which may contribute to fractures), muscular weakness, myopathy.
Renal and Urinary Disorders: Acute renal failure, renal failure, acute tubular necrosis, Fanconi syndrome, proximal renal tubulopathy, interstitial nephritis (including acute cases), nephrogenic diabetes insipidus, renal insufficiency, increased creatinine, proteinuria, polyuria.
General Disorders and Administration Site Conditions: Asthenia.
The following adverse reactions, listed under the body system headings above, may occur as a consequence of proximal renal tubulopathy: rhabdomyolysis, osteomalacia, hypokalemia, muscular weakness, myopathy, hypophosphatemia.
View ADR Monitoring Form