


 Sign Out
Sign Out

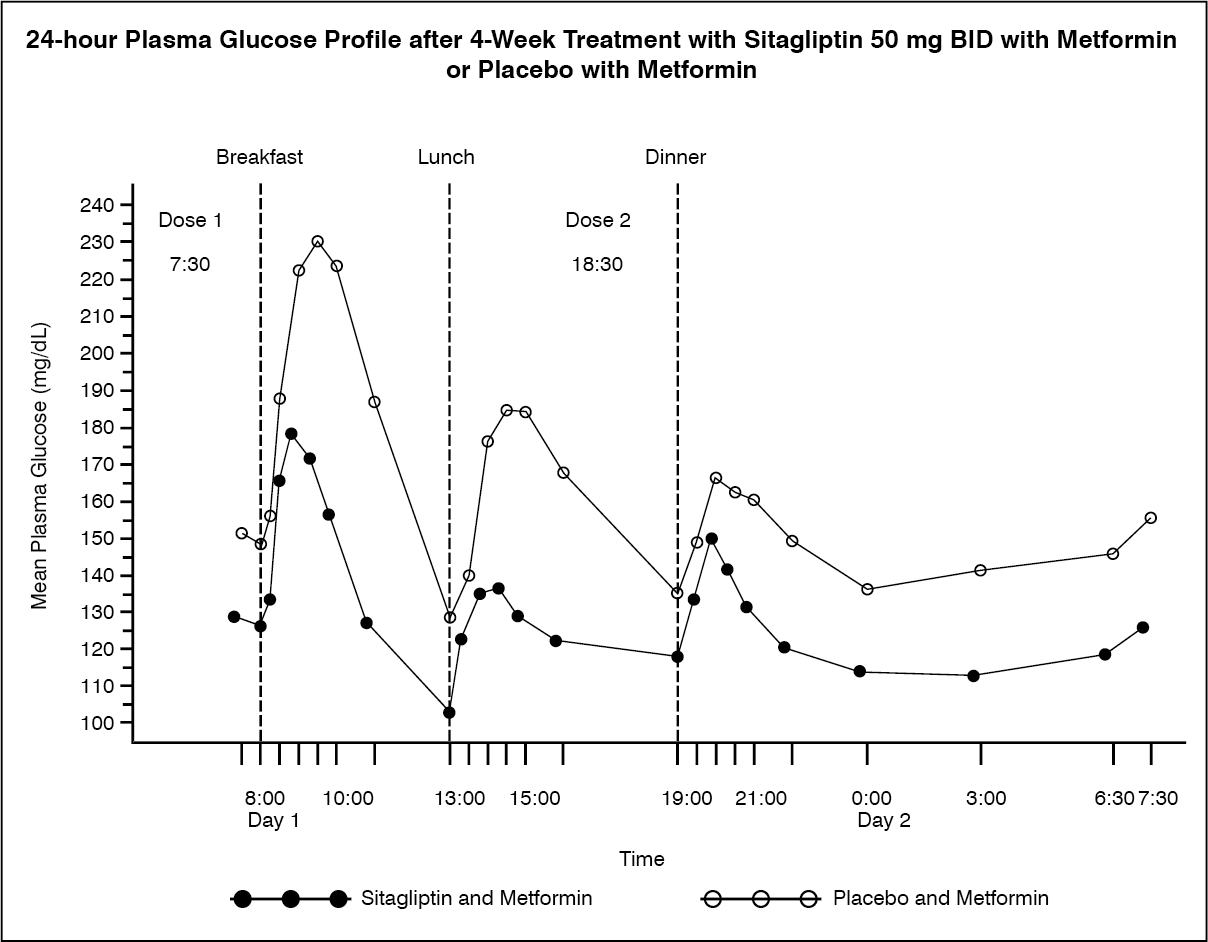
In a study of patients with type 2 diabetes inadequately controlled on metformin monotherapy, glucose levels monitored throughout the day were significantly lower in patients who received sitagliptin 100 mg per day (50 mg twice daily) in combination with metformin compared with patients who received placebo with metformin (see Figure).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn Phase III clinical studies of 18- and 24-week duration, treatment with SITAGLIPTIN (XELEVIA) 100 mg daily in patients with type 2 diabetes significantly improved beta cell function, as assessed by several markers, including HOMA-β (Homeostasis Model Assessment-β), proinsulin to insulin ratio, and measures of beta cell responsiveness from the frequently-sampled meal tolerance test.
In Phase II studies, SITAGLIPTIN (XELEVIA) 50 mg twice daily provided no additional glycemic efficacy compared to 100 mg once daily.
In a randomized, placebo-controlled, double-blind, double-dummy, four-period crossover study in healthy adult subjects, the effects on post-meal plasma concentrations of active and total GLP-1 and glucose after co-administration of sitagliptin and metformin were compared with those after administration of sitagliptin alone, metformin alone, or placebo, each administered for two days. The incremental 4-hour post-meal weighted mean active GLP-1 concentrations were increased by approximately 2-fold after either administration of sitagliptin alone or metformin alone compared with placebo. The effect on active GLP-1 concentrations after co-administration of sitagliptin and metformin were additive, with active GLP-1 concentrations increased by approximately 4-fold compared with placebo. Sitagliptin alone increased only active GLP-1 concentrations, reflecting inhibition of DPP-4, whereas metformin alone increased active and total GLP-1 concentrations to a similar extent. These data are consistent with different mechanisms for the increase in active GLP-1 concentrations. Results from the study also demonstrated that sitagliptin, but not metformin, enhances active GIP concentrations.
In studies with healthy subjects, SITAGLIPTIN (XELEVIA) did not lower blood glucose or cause hypoglycemia, suggesting that the insulinotropic and glucagon suppressive actions of the drug are glucose dependent.
Effects on blood pressure: In a randomized, placebo-controlled crossover study in hypertensive patients on one or more anti-hypertensive drugs (including angiotensin-converting enzyme inhibitors, angiotensin-II antagonists, calcium-channel blockers, beta-blockers and diuretics), co-administration with SITAGLIPTIN (XELEVIA) was generally well tolerated. In these patients, SITAGLIPTIN (XELEVIA) had a modest blood pressure lowering effect; 100 mg per day of SITAGLIPTIN (XELEVIA) reduced 24-hour mean ambulatory systolic blood pressure by approximately 2 mmHg, as compared to placebo. Reductions have not been observed in subjects with normal blood pressure.
Cardiac Electrophysiology: In a randomized, placebo-controlled crossover study, 79 healthy subjects were administered a single oral dose of SITAGLIPTIN (XELEVIA) 100 mg, SITAGLIPTIN (XELEVIA) 800 mg (8 times the recommended dose), and placebo. At the recommended dose of 100 mg, there was no effect on the QTc interval obtained at the peak plasma concentration, or at any other time during the study. Following the 800-mg dose, the maximum increase in the placebo-corrected mean change in QTc from baseline at 3 hours postdose was 8.0 msec. This small increase was not considered to be clinically significant. At the 800-mg dose, peak sitagliptin plasma concentrations were approximately 11 times higher than the peak concentrations following a 100 mg dose.
In patients with type 2 diabetes administered SITAGLIPTIN (XELEVIA) 100 mg (N=81) or SITAGLIPTIN (XELEVIA) 200 mg (N=63) daily, there were no meaningful changes in QTc interval based on ECG data obtained at the time of expected peak plasma concentration.
Pharmacokinetics: The pharmacokinetics of sitagliptin have been extensively characterized in healthy subjects and patients with type 2 diabetes. After oral administration of a 100-mg dose to healthy subjects, sitagliptin was rapidly absorbed, with peak plasma concentrations (median Tmax) occurring 1 to 4 hours post‑dose. Plasma AUC of sitagliptin increased in a dose-proportional manner. Following a single oral 100-mg dose to healthy volunteers, mean plasma AUC of sitagliptin was 8.52 μM·hr, Cmax was 950 nM, and apparent terminal half-life (t1/2) was 12.4 hours. Plasma AUC of sitagliptin increased approximately 14% following 100-mg doses at steady-state compared to the first dose. The intra-subject and inter-subject coefficients of variation for sitagliptin AUC were small (5.8% and 15.1%). The pharmacokinetics of sitagliptin were generally similar in healthy subjects and in patients with type 2 diabetes.
Absorption: The absolute bioavailability of sitagliptin is approximately 87%. Since co-administration of a high-fat meal with SITAGLIPTIN (XELEVIA) had no effect on the pharmacokinetics, SITAGLIPTIN (XELEVIA) may be administered with or without food.
Distribution: The mean volume of distribution at steady state following a single 100-mg intravenous dose of sitagliptin to healthy subjects is approximately 198 liters. The fraction of sitagliptin reversibly bound to plasma proteins is low (38%).
Metabolism: Sitagliptin is primarily eliminated unchanged in urine, and metabolism is a minor pathway. Approximately 79% of sitagliptin is excreted unchanged in the urine.
Following a [14C] sitagliptin oral dose, approximately 16% of the radioactivity was excreted as metabolites of sitagliptin. Six metabolites were detected at trace levels and are not expected to contribute to the plasma DPP-4 inhibitory activity of sitagliptin. In vitro studies indicated that the primary enzyme responsible for the limited metabolism of sitagliptin was CYP3A4, with contribution from CYP2C8.
Elimination: Following administration of an oral [14C] sitagliptin dose to healthy subjects, approximately 100% of the administered radioactivity was eliminated in feces (13%) or urine (87%) within one week of dosing. The apparent terminal t1/2 following a 100-mg oral dose of sitagliptin was approximately 12.4 hours and renal clearance was approximately 350 mL/min.
Elimination of sitagliptin occurs primarily via renal excretion and involves active tubular secretion. Sitagliptin is a substrate for human organic anion transporter-3 (hOAT-3), which may be involved in the renal elimination of sitagliptin. The clinical relevance of hOAT-3 in sitagliptin transport has not been established. Sitagliptin is also a substrate of p-glycoprotein, which may also be involved in mediating the renal elimination of sitagliptin. However, cyclosporine, a p-glycoprotein inhibitor, did not reduce the renal clearance of sitagliptin.
Characteristics in Patients: Renal Impairment: A single-dose, open-label study was conducted to evaluate the pharmacokinetics of SITAGLIPTIN (XELEVIA) (50 mg dose) in patients with varying degrees of chronic renal impairment compared to normal healthy control subjects. The study included patients with mild, moderate, and severe renal impairment, as well as patients with ESRD on hemodialysis. In addition, the effects of renal impairment on sitagliptin pharmacokinetics in patients with type 2 diabetes and mild, moderate or severe renal impairment (including ESRD) were assessed using population pharmacokinetic analyses.
Compared to normal healthy control subjects, plasma AUC of sitagliptin was increased by approximately 1.2-fold and 1.6-fold in patients with mild renal impairment (eGFR ≥ 60 mL/min/1.73 m2 to < 90 mL/min/1.73 m2) and patients with moderate renal impairment (eGFR ≥ 45 mL/min/1.73 m2 to < 60 mL/min/1.73 m2), respectively. Because increases of this magnitude are not clinically relevant, dosage adjustment in these patients is not necessary.
Plasma AUC of sitagliptin was increased approximately 2-fold in patients with moderate renal impairment (eGFR ≥ 30 mL/min/1.73 m2 to < 45 mL/min/1.73 m2), and approximately 4-fold in patients with severe renal impairment (eGFR < 30 mL/min/1.73 m2), including patients with ESRD on hemodialysis. Sitagliptin was modestly removed by hemodialysis (13.5% over a 3- to 4-hour hemodialysis session starting 4 hours post-dose). To achieve plasma concentrations of sitagliptin similar to those in patients with normal renal function, lower dosages are recommended in patients with eGFR <45 mL/min/1.73 m2. (See Patients with Renal Impairment under Dosage & Administration.)
Hepatic Impairment: In patients with moderate hepatic impairment (Child-Pugh score 7 to 9), mean AUC and Cmax of sitagliptin increased approximately 21% and 13%, respectively, compared to healthy matched controls following administration of a single 100 mg dose of SITAGLIPTIN (XELEVIA). These differences are not considered to be clinically meaningful. No dosage adjustment for SITAGLIPTIN (XELEVIA) is necessary for patients with mild or moderate hepatic impairment.
There is no clinical experience in patients with severe hepatic impairment (Child-Pugh score >9). However, because sitagliptin is primarily renally eliminated, severe hepatic impairment is not expected to affect the pharmacokinetics of sitagliptin.
Elderly: No dosage adjustment is required based on age. Age did not have a clinically meaningful impact on the pharmacokinetics of sitagliptin based on a population pharmacokinetic analysis of Phase I and Phase II data. Elderly subjects (65 to 80 years) had approximately 19% higher plasma concentrations of sitagliptin compared to younger subjects.
Pediatric: The pharmacokinetics of sitagliptin (single dose of 50 mg, 100 mg or 200 mg) were investigated in pediatric patients (10 to 17 years of age) with type 2 diabetes. In this population, the dose-adjusted AUC of sitagliptin in plasma was approximately 18% lower compared to adult patients with type 2 diabetes for a 100 mg dose. This is not considered to be a clinically meaningful difference based on the flat PK/PD relationship between the dose of 50 mg and 100 mg in adults.
No studies with sitagliptin have been performed in pediatric patients < 10 years of age.
Gender: No dosage adjustment is necessary based on gender. Gender had no clinically meaningful effect on the pharmacokinetics of sitagliptin based on a composite analysis of Phase I pharmacokinetic data and on a population pharmacokinetic analysis of Phase I and Phase II data.
Race: No dosage adjustment is necessary based on race. Race had no clinically meaningful effect on the pharmacokinetics of sitagliptin based on a composite analysis of Phase I pharmacokinetic data and on a population pharmacokinetic analysis of Phase I and Phase II data, including subjects of white, Hispanic, black, Asian, and other racial groups.
Body Mass Index (BMI): No dosage adjustment is necessary based on BMI. Body mass index had no clinically meaningful effect on the pharmacokinetics of sitagliptin based on a composite analysis of Phase I pharmacokinetic data and on a population pharmacokinetic analysis of Phase I and Phase II data.
Type 2 Diabetes: The pharmacokinetics of sitagliptin in patients with type 2 diabetes are generally similar to those in healthy subjects.