Each film-coated tablet contains: Sitagliptin (as phosphate) 50 mg.
Each film-coated tablet contains: Sitagliptin (as phosphate) 100 mg.
Sitagliptin (Xiglip) tablets contain Sitagliptin phosphate, an orally-active inhibitor of the dipeptidyl peptidase-4 (DPP-4) enzyme. Sitagliptin phosphate monohydrate is described chemically as 7-[(3R)-3-amino-1-oxo-4-(2,4,5-trifluorophenyl) butyl]-5,6,7,8-tetrahydro-3-(trifluoromethyl)-1,2,4-triazolo[4,3-a]pyrazine phosphate (1:1) monohydrate.
The empirical formula is C16H15F6N5O.H3PO4 and the molecular weight is 505.32.
Sitagliptin phosphate is a white to off-white powder. Each tablet of sitagliptin contains 31.015, 62.031, or 124.061 mg of Sitagliptin phosphate, which is equivalent to 50, or 100 mg, respectively, of free base.
Excipients/Inactive ingredients: Microcrystalline cellulose, anhydrous dibasic calcium phosphate, croscarmellose sodium, magnesium stearate, and sodium stearyl fumarate.
Film coating: polyvinyl alcohol, polyethylene glycol, talc, titanium dioxide, red iron oxide and yellow iron oxide.
Pharmacotherapeutic group: Drugs used in diabetes, Dipeptidyl peptidase4 (DPP-4) inhibitors. ATC code: A10BH01.
Pharmacology: Pharmacodynamics: Mechanism of action: Sitagliptin is a member of a class of oral anti-hyperglycemic agents called dipeptidyl peptidase 4 (DPP-4) inhibitors. The
improvement in glycemic control observed with this medicinal product may be mediated by enhancing the levels of active incretin hormones. Incretin hormones, including glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), are released by the intestine throughout the day, and levels are increased in response to a meal. The incretins are part of an endogenous system involved in the physiologic regulation of glucose homeostasis. When blood glucose concentrations are normal or elevated, GLP-1 and GIP increase insulin synthesis and release from pancreatic beta cells by intracellular signaling pathways involving cyclic AMP. Treatment with GLP-1 or with DPP-4 inhibitors in animal models of type 2 diabetes has been demonstrated to improve beta cell responsiveness to glucose and stimulate insulin biosynthesis and release. With higher insulin levels, tissue glucose uptake is enhanced. In addition, GLP-1 lowers glucagon secretion from pancreatic alpha cells. Decreased glucagon concentrations, along with higher insulin levels, lead to reduced hepatic glucose production, resulting in a decrease in blood glucose levels. The effects of GLP-1 and GIP are glucose-dependent such that when blood glucose concentrations are low, stimulation of insulin release and suppression of glucagon secretion by GLP-1 are not observed. For both GLP-1 and GIP, stimulation of insulin release is enhanced as glucose rises above normal concentrations. Further, GLP-1 does not impair the normal glucagon response to hypoglycemia. The activity of GLP-1 and GIP is limited by the DPP-4 enzyme, which rapidly hydrolyzes the incretin hormones to produce inactive products. Sitagliptin prevents the hydrolysis of incretin hormones by DPP-4, thereby increasing plasma concentrations of the active forms ofGLP-1 and GIP. By enhancing active incretin levels, sitagliptin increases insulin release and decreases glucagon levels in a glucose-dependent manner. In patients with type 2 diabetes with hyperglycemia, these changes in insulin and glucagon levels lead to lower hemoglobin A1c (HbA1c) and lower fasting and postprandial glucose concentrations. The glucose-dependent mechanism of sitagliptin is distinct from the mechanism of sulphonylureas, which increase insulin secretion even when glucose levels are low and can lead to hypoglycemia in patients with type 2 diabetes and in normal subjects. Sitagliptin is a potent and highly selective inhibitor of the enzyme DPP-4 and does not inhibit the closely-related enzymes DPP-8 or DPP-9 at therapeutic concentrations. In a two-day study in healthy subjects, sitagliptin alone increased active GLP-1 concentrations, whereas metformin alone increased active and total GLP-1 concentrations to similar extents. Co-administration of sitagliptin and metformin had an additive effect on active GLP-1 concentrations. Sitagliptin, but not metformin, increased active GIP concentrations.
Pharmacokinetics: Absorption: Following oral administration of a 100 mg dose to healthy subjects, sitagliptin was rapidly absorbed, with peak plasma concentrations (median Tmax) occurring 1 to 4 hours post-dose, mean plasma AUC of sitagliptin was 8.52 μMhr, Cmax as 950 nM. The absolute bioavailability of sitagliptin is approximately 87%. Since co-administration of a high fat meal with sitagliptin had no effect on the pharmacokinetics, sitagliptin may be administered with or without food. Plasma AUC of sitagliptin increased in a dose-proportional manner. Dose proportionality was not established for Cmax and C24hr (Cmax increased in a greater than dose-proportional manner and C24hrincreased in a less than dose-proportional manner).
Distribution: The mean volume of distribution at steady state following a single 100 mg intravenous dose of sitagliptin to healthy subjects is approximately 198 liters. The fraction of sitagliptin reversibly bound to plasma proteins is low (38%).
Biotransformation: Sitagliptin is primarily eliminated unchanged in urine, and metabolism is a minor pathway. Approximately 79% of sitagliptin is excreted unchanged in the urine. Following a [14C] sitagliptin oral dose, approximately 16% of the radioactivity was excreted as metabolites of sitagliptin. Six metabolites were detected at trace levels and are not expected to contribute to the plasma DPP-4 inhibitory activity of sitagliptin. In vitro studies indicated that the primary enzyme responsible for the limited metabolism of sitagliptin was CYP3A4, with contribution from CYP2C8. In vitro data showed that sitagliptin is not an inhibitor of CYP isozymes CYP3A4, 2C8, 2C9, 2D6, 1A2, 2C19 or2B6, and is not an inducer of CYP3A4 and CYP1A2.
Elimination: Following administration of an oral [14C] sitagliptin dose to healthy subjects, approximately 100% of the administered
radioactivity was eliminated in feces (13%) or urine (87%) within one week of dosing. The apparent terminal t1/2 following a 100
mg oral dose of sitagliptin was approximately 12.4 hours. Sitagliptin accumulates only minimally with multiple doses. The renal
clearance was approximately 350 ml/min. Elimination of sitagliptin occurs primarily via renal excretion and involves active
tubular secretion. Sitagliptin is a substrate for human organic anion transporter-3 (hOAT-3), which may be involved in the renal
elimination of sitagliptin. The clinical relevance of hOAT-3 in sitagliptin transport has not been established. Sitagliptin is also a
substrate of p-glycoprotein, which may also be involved in mediating the renal elimination of sitagliptin. However, ciclosporin, a p-glycoprotein inhibitor, did not reduce the renal clearance of sitagliptin. Sitagliptin is not a substrate for OCT2 or OAT1 or PEPT1/2 transporters. In vitro, sitagliptin did not inhibit OAT3 (IC50=160 μM) or p-glycoprotein (up to 250 μM) mediated transport at therapeutically relevant plasma concentrations. In a clinical study sitagliptin had a small effect on plasma digoxin concentrations indicating that sitagliptin may be a mild inhibitor of p-glycoprotein.
Specific Patients: The pharmacokinetics of sitagliptin were generally similar in healthy subjects and in patients with type 2 diabetes.
Renal Impairment: A single dose, open-label study was conducted to evaluate the pharmacokinetics of a reduced dose of sitagliptin (50 mg) in patients with varying degrees of chronic renal impairment compared to normal healthy control subjects. The study included patients with renal impairment classified on the basis of creatinine clearance as mild (50 to <80 ml/min), moderate (30 to <50 ml/min), and severe (< 30 ml/min), as well as patients with end-stage renal disease (ESRD) on hemodialysis. Patients with mild
renal impairment did not have a clinically meaningful increase in the plasma concentration of sitagliptin as compared to normal
healthy control subjects. An approximately 2-fold increase in the plasma AUC of sitagliptin was observed in patients with
moderate renal impairment, and an approximately 4-fold increase was observed in patients with severe renal impairment and in
patients with ESRD on hemodialysis, as compared to normal healthy control subjects. Sitagliptin was modestly removed by
hemodialysis (13.5% over a 3- to 4-hourhemodialysis session starting 4 hours post-dose). To achieve plasma concentrations of
sitagliptin similar to those in patients with normal renal function, lower dosages are recommended in patients with moderate and
severe renal impairment, as well as in ESRD patients requiring dialysis.
Hepatic Impairment: No dose adjustment for sitagliptin is necessary for patients with mild or moderate hepatic impairment (Child-Pugh scores ≤9). There is no clinical experience in patients with severe hepatic impairment (Child-Pugh score >9). However, because sitagliptin is primarily renally eliminated, severe hepatic impairment is not expected to affect the pharmacokinetics of sitagliptin.
Geriatric: No dose adjustment is required based on age. Age did not have a clinically meaningful impact on the pharmacokinetics of
sitagliptin based on a population pharmacokinetic analysis. Elderly subjects (65 to 80 years) had approximately 19% higher plasma concentrations of sitagliptin compared to younger subjects.
Pediatric: No studies with sitagliptin have been performed in pediatric patients.
Other patients: No dose adjustment is necessary based on gender, race, or body mass index (BMI). These characteristics had no clinically meaningful effect on the pharmacokinetics of sitagliptin based on a composite analysis of Phase I pharmacokinetic data and on a population pharmacokinetic analysis of Phase I and Phase II data.
For adult patients with type 2 diabetes mellitus, sitagliptin is indicated to improve glycemic control: As monotherapy: In patients inadequately controlled by diet and exercise alone and for whom metformin is inappropriate due to contraindications or intolerance.
As dual oral therapy in combination with: Metformin when diet and exercise plus metformin alone do not provide adequate glycemic control. A sulphonylureas when diet and exercise plus maximal tolerated dose of a sulphonylurea alone do not provide adequate glycemic control and when metformin is inappropriate due to contraindications or intolerance.
A peroxisome proliferator-activated receptor gamma (PPARγ) agonist (i.e. a thiazolidinedione)when use of a PPARγ agonist is appropriate and when diet and exercise plus the PPARγ agonist alone do not provide adequate glycemic control.
As triple oral therapy in combination with: A sulphonylureas and metformin when diet and exercise plus dual therapy with these medicinal products do not provide adequate glycemic control.
A PPARγ agonist and metformin when use of a PPARγ agonist is appropriate and when diet and exercise plus dual therapy with these medicinal products do not provide adequate glycaemic control.
Sitagliptin is also indicated as add-on to insulin (with or without metformin) when diet and exercise plus stable dose of insulin do not provide adequate glycemic control.
The dose is 100 mg sitagliptin once daily. When used in combination with metformin and/or a PPARγ agonist, the dose of metformin and/or PPARγ agonist should be maintained, and sitagliptin administered concomitantly.
When sitagliptin is used in combination with a sulphonylureas or with insulin, a lower dose of the sulphonylureas or insulin may
be considered to reduce the risk of hypoglycemia. If a dose of sitagliptin is missed, it should be taken as soon as the patient
remembers. A double dose should not be taken on the same day.
Recommended Dose for Specific Population: Renal Impairment: Patients with an estimated glomerular filtration rate [eGFR] greater than or equal to 45 mL/min/1.73 m2 to less than 90
mL/min/1.73 m2, no dosage adjustment for Sitagliptin is required.
Patients with moderate renal impairment (eGFR greater than or equal to 30 mL/min/1.73 m2 to less than 45 ml/min/1.73 m2),
the dose of sitagliptin is 50 mg once daily.
Patients with severe renal impairment (eGFR less than 30 mL/min/1.73 m2) or with end-stage renal disease (ESRD) requiring
hemodialysis or peritoneal dialysis, the dose of sitagliptin is 25 mg once daily.
Sitagliptin may be administered without regard to the timing of dialysis. Assess renal function prior to initiation of sitagliptin and
periodically thereafter.
Hepatic Impairment: No dose adjustment is necessary for patients with mild to moderate hepatic impairment. Sitagliptin has not been studied in
patients with severe hepatic impairment and care should be exercised. However, because sitagliptin is primarily renally
eliminated, severe hepatic impairment is not expected to affect the pharmacokinetics of sitagliptin.
Geriatrics:
In pre-approval clinical safety and efficacy studies of sitagliptin, 725 patients were 65 years and over, while 61 patients were 75
years and over. No overall differences in safety or effectiveness were observed between subjects 65 years and over and younger
subjects. While this and other reported clinical experience have not identified differences in responses between the elderly and
younger patients, greater sensitivity of some older individuals cannot be ruled out. Sitagliptin is substantially excreted by the
kidney. Aging can be associated with reduced renal function, therefore, elderly patients should be assessed and monitored more
frequently.
Pediatric Population:
The safety and efficacy of sitagliptin in children and adolescents under 18 years of age have not yet been established.
Method of Administration:
Sitagliptin can be taken with or without food.
Oral route.
During controlled clinical trials in healthy subjects, single doses of up to 800 mg sitagliptin were administered. Minimal increases in QTc, not considered to be clinically relevant, were observed in one study at a dose of 800 mg sitagliptin. There is no experience with doses above 800 mg in clinical studies. In Phase I multiple dose studies, there were no dose-related clinical adverse reactions observed with sitagliptin with doses of up to 600 mg per day for periods of up to 10 days and 400 mg per day for periods of up to 28 days. In the event of an overdose, it is reasonable to employ the usual supportive measures, e.g., remove unabsorbed material from the gastrointestinal tract, employ clinical monitoring (including obtaining an electrocardiogram), and institute supportive therapy if required.
Sitagliptin is modestly dialyzable. In clinical studies, approximately 13.5% of the dose was removed over a 3- to 4-hour hemodialysis session. Prolonged hemodialysis may be considered if clinically appropriate. It is not known if sitagliptin is dialyzable by peritoneal dialysis.
Hypersensitivity to the active substance or to any of its excipients.
General: Sitagliptin should not be used in patients with type 1 diabetes or for the treatment of diabetic ketoacidosis.
Acute pancreatitis: Use of DPP-4 inhibitors has been associated with a risk of developing acute pancreatitis. Patients should be informed of the characteristic symptom of acute pancreatitis: persistent, severe abdominal pain. Resolution of pancreatitis has been observed after discontinuation of sitagliptin (with or without supportive treatment), but very rare cases of necrotising or hemorrhagic pancreatitis and/or death have been reported. If pancreatitis is suspected, sitagliptin and other potentially suspect medicinal products should be discontinued; if acute pancreatitis is confirmed, sitagliptin should not be restarted.
Caution should be exercised in patients with a history of pancreatitis.
Hypoglycemia with Concomitant Use with Other Anti-Hyperglycemic Medicinal Products: In clinical trials of sitagliptin as monotherapy and as part of combination therapy with medicinal products not known to cause hypoglycemia (i.e. metformin and/or a PPARγ agonist), rates of hypoglycemia reported with sitagliptin were similar to rates in patients taking placebo. Hypoglycemia has been observed when sitagliptin was used in combination with insulin or a sulphonylureas. Therefore, to reduce the risk of hypoglycemia, a lower dose of sulphonylureas or insulin may be considered.
Heart Failure: Consider the risks and benefits of sitagliptin prior to initiating treatment in patients at risk for heart failure, such as those with a prior history of heart failure and observe these patients for signs and symptoms of heart failure during therapy.
Advise patients to immediately report such symptoms. If heart failure develops, evaluate and manage the case according to current procedures and standards of care and consider discontinuation of sitagliptin.
Renal Impairment: Sitagliptin is renally excreted. To achieve plasma concentrations of sitagliptin similar to those in patients with normal renal function, lower dosages are recommended in patients with moderate and severe renal impairment, as well as in ESRD patients requiring hemodialysis or peritoneal dialysis. When considering the use of sitagliptin in combination with another anti-diabetic medicinal product, its conditions for use in patients with renal impairment should be checked.
Hypersensitivity Reactions: Post-marketing reports of serious hypersensitivity reactions in patients treated with sitagliptin have been reported. These reactions include anaphylaxis, angioedema, and exfoliative skin conditions including Stevens-Johnson syndrome. Onset of these reactions occurred within the first 3 months after initiation of treatment, with some reports occurring after the first dose. If a hypersensitivity reaction is suspected, sitagliptin should be discontinued. Other potential causes for the event should be assessed, and alternative treatment for diabetes initiated.
Severe and Disabling Arthralgia: There have been post-marketing reports of severe and disabling arthralgia in patients taking DPP-4 inhibitors. The time to onset of symptoms following initiation of drug therapy varied from one day to years. Patients experienced relief of symptoms upon discontinuation of the medication. A subset of patients experienced a recurrence of symptoms when restarting the same drug or a different DPP-4 inhibitor. Consider DPP-4 inhibitors as a possible cause for severe joint pain.
Bullous Pemphigoid: Post-marketing cases of bullous pemphigoid requiring hospitalization have been reported with DPP-4 inhibitor use. In reported cases, patients typically recovered with topical or systemic immunosuppressive treatment and discontinuation of the DPP-4 inhibitor. Patient should report development of blisters or erosions while receiving Sitagliptin. If bullous pemphigoid is suspected, sitagliptin should be discontinued and refer the patient to a dermatologist for diagnosis and appropriate treatment.
Pregnancy: Category B.
The limited available data with sitagliptin in pregnant women are not sufficient to inform a drug-associated risk for major birth defects and miscarriage. There are risks to the mother and fetus associated with poorly controlled diabetes in pregnancy (see Clinical Considerations as follows). No adverse developmental effects were observed when sitagliptin was administered to pregnant rats and rabbits during organogenesis at oral doses up to 30-times and 20-times, respectively, the 100 mg clinical dose, based on AUG (see Data as follows).
The estimated background risk of major birth defects is 6-10% in women with pre-gestational diabetes with a Hemoglobin A1c >7% and has been reported to be as high as 20-25% in women with a Hemoglobin A1c >10%. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations: Maternal and/or Fatal Risk: Poorly controlled diabetes in pregnancy increases the maternal risk for diabetic ketoacidosis, pre-eclampsia, spontaneous abortions, preterm delivery, and delivery complications. Poorly controlled diabetes increases the fetal risk for major birth defects, still birth, and macrosomia related morbidity.
Data: In embryo-fetal development studies, sitagliptin administered to pregnant rats and rabbits during organogenesis (gestation day 6 to 20) did not adversely affect developmental outcomes at oral doses up to 250 mg/kg (30-times the 100 mg clinical dose) and 125 mg/kg (20-times the 100 mg clinical dose), respectively, based on AUC. Higher doses in rats associated with maternal toxicity increased the incidence of rib malformations in offspring at 1000 mg/kg, or approximately 100-times the clinical dose, based on AUC. Placental transfer of sitagliptin was observed in pregnant.
Lactation: Sitagliptin is present in rat milk. It is secreted in the milk of lactating rats at a milk to plasma ratio of 4:1. Therefore, possibly present in human milk. It is unknown whether sitagliptin excreted in human breast milk and due to lack of human data, sitagliptin should not be used during breastfeeding.
Fertility: In rat fertility studies with oral gavage doses of 125, 250, and 1000 mg/kg, males were treated for 4 weeks prior to mating, during mating, up to scheduled termination (approximately 8 weeks total) and females were treated 2 weeks prior to mating through gestation day 7. No adverse effect on fertility was observed at 125 mg/kg (approximately 12 times human exposure at the MRHD of 100 mg/day based on AUC comparisons). At higher doses, non-dose related increased resorptions in females were observed (approximately 25 and 100 times human exposure at the MRHD based on AUC comparison).
No human fertility studies.
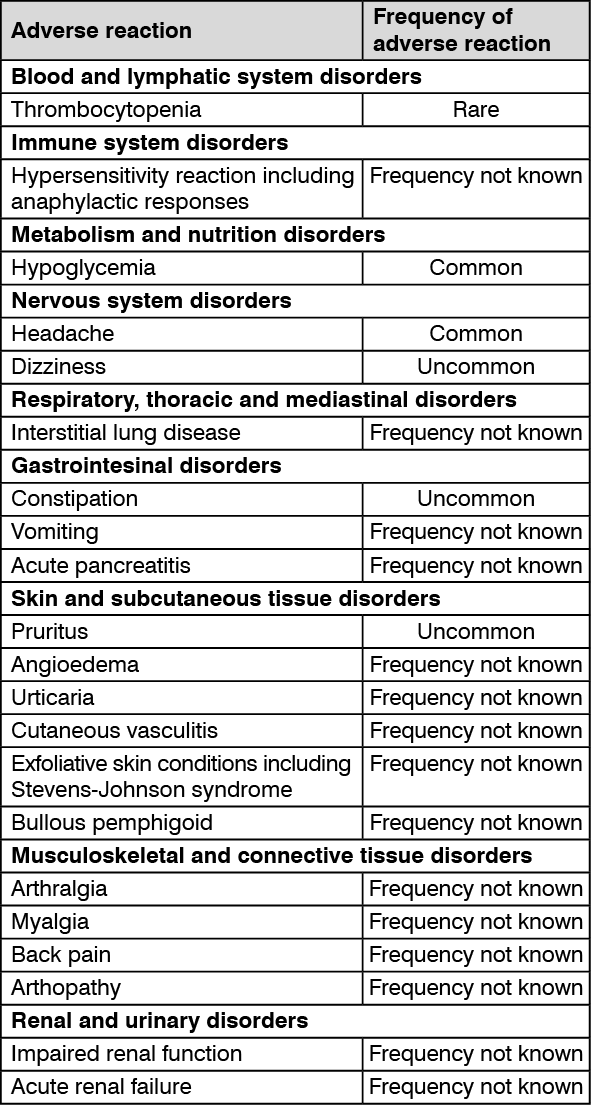
Adverse reactions are listed as follows by system organ class and frequency. Frequencies are defined as: very common (≥ 1/10);
common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000) and not known (cannot be estimated from the available data).
The frequency of adverse reactions identified from placebo-controlled clinical studies of sitagliptin monotherapy and post-marketing experience: See table.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Effects of Other Medicinal Products on Sitagliptin: Clinical data described as follows suggest that the risk for clinically meaningful interactions by co-administered medicinal products is low. In vitro studies indicated that the primary enzyme responsible for the limited metabolism of sitagliptin is CYP3A4, with contribution from CYP2CB. In patients with normal renal function, metabolism, including via CYP3A4, plays only a small role in the clearance of sitagliptin. Metabolism may play a more significant role in the elimination of sitagliptin in the setting of severe renal impairment or end-stage renal disease (ESRD). For this reason, it is possible that potent CYP3A4 inhibitors (i.e. ketoconazole, itraconazole, ritonavir, clarithromycin) could alter the pharmacokinetics of sitagliptin in patients with severe renal impairment or ESRD.
The effect of potent CYP3A4 inhibitors in the setting of renal impairment has not been assessed in a clinical study. In vitro transport studies showed that sitagliptin is a substrate for p-glycoprotein and organic anion transporter-3 (OAT3). OAT3 mediated transport of sitagliptin was inhibited in vitro by probenecid, although the risk of clinically meaningful interactions is considered to be low. Concomitant administration of OAT3 inhibitors has not been evaluated in vitro.
Metformin: Co-administration of multiple twice-daily doses of 1,000 mg metformin with 50 mg sitagliptin did not meaningfully alter the pharmacokinetics of sitagliptin in patients with type 2 diabetes.
Ciclosporin: A study was conducted to assess the effect of ciclosporin, a potent inhibitor of p-glycoprotein, on the pharmacokinetics of sitagliptin. Co-administration of a single 100 mg oral dose of sitagliptin and a single 600 mg oral dose of ciclosporin increased the AUC and Cmax of sitagliptin by approximately 29% and 68%, respectively. These changes in sitagliptin pharmacokinetics were not considered to be clinically meaningful. The renal clearance of sitagliptin was not meaningfully altered. Therefore, meaningful interactions would not be expected with other p-glycoprotein inhibitors.
Effects of Sitagliptin on Other Medicinal Products: Digoxin: Sitagliptin had a small effect on plasma digoxin concentrations. Following administration of 0.25 mg digoxin concomitantly with 100 mg of sitagliptin daily for 10 days, the plasma AUC of digoxin was increased on average by 11%, and the plasma Cmax on average by 18%. No dose adjustment of digoxin is recommended. However, patients at risk of digoxin toxicity should be monitored forth is when sitagliptin and digoxin are administered concomitantly.
In vitro data suggest that sitagliptin does not inhibit nor induce CYP450 isoenzymes. In clinical studies, sitagliptin did not meaningfully alter the pharmacokinetics of metformin, glyburide, simvastatin, rosiglitazone, warfarin, or oral contraceptives, providing in vivo evidence of a low propensity for causing interactions with substrates of CYP3A4, CYP2C8, CYP2C9, and organic cationic transporter (OCT). Sitagliptin may be a mild inhibitor of p-glycoprotein in vivo.
Instructions and Special Precautions for Handling and Disposal: There is no special instructions for handling and disposal of this medicine.
Store at temperatures not exceeding 30°C.
Do not store in direct sunlight or heat.
A10BH01 - sitagliptin ; Belongs to the class of dipeptidyl peptidase 4 (DPP-4) inhibitors. Used in the treatment of diabetes.
Xiglip FC tab 100 mg
30's


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out