Commonly Observed: The most commonly observed adverse experiences associated with the use of paroxetine in clinical trials and not seen at an equivalent incidence among placebo-treated patients were: nausea, somnolence, sweating, tremor, asthenia, dizziness, dry mouth, insomnia, constipation, diarrhea, decreased appetite and male sexual dysfunction (see Tables 3 and 4).
Adverse Events Leading to Discontinuation of Treatment: Twenty-one percent of over 4000 patients who received paroxetine in worldwide clinical trials in depression discontinued treatment due to an adverse experience. In obsessive-compulsive disorder, panic disorder, social phobia (social anxiety disorder), generalized anxiety disorder and posttraumatic stress disorder studies, 11.8% (64/542), 9.4% (44/469), 16.1% (84/522), 10.7% (79/735) and 11.7% (79/676), respectively, of patients treated with paroxetine discontinued treatment because of adverse events. The most common events leading to discontinuation (reported by 1% or more of subjects) included: asthenia, headache, nausea, somnolence, insomnia, agitation, tremor, dizziness, constipation, impotence, abnormal ejaculation, sweating and diarrhea.
Adverse Effects Following Discontinuation of Treatment (or Dose Reduction) Clinical Trials: The following adverse events have been reported at an incidence of 2% or greater for paroxetine and were at least twice that reported for placebo: abnormal dreams (2.3% vs 0.5%), paresthesias (2.0% vs 0.4%), and dizziness (7.1% vs 1.5%). The majority of these events were mild to moderate, self-limiting and did not require medical intervention. These adverse events were noted in GAD and PTSD clinical trials employing a taper phase regimen for discontinuation of treatment.
This regimen involved an incremental decrease in the daily dose by 10 mg/day at weekly intervals. When a daily dose of 20 mg/day was reached, patients were continued on this dose for 1 week before treatment was stopped.
Post-Marketing: There have been spontaneous reports of adverse events upon the discontinuation of paroxetine (particularly when abrupt), including but not limited to the following: dizziness, sensory disturbances (including paresthesias and electric shock sensations), agitation/restlessness, anxiety, nausea, vomiting, sweating, headache, and sleep disturbance. These events are generally self-limiting. Symptoms associated with discontinuation have been reported for other selective serotonin reuptake inhibitors.
Patients should be monitored for these or any other symptoms when discontinuing treatment, regardless of the indication for which paroxetine is being prescribed. If intolerable symptoms occur following a decrease in the dose or upon discontinuation of treatment, dose titration should be managed on the basis of the patient's clinical response (see PRECAUTIONS and DOSAGE & ADMINISTRATION).
Clinical Trial Experience: Multiple doses of paroxetine were administered to 4126 subjects in clinical trials for depression, 542 subjects in clinical trials for OCD, 469 subjects in clinical trials for panic disorder, 522 subjects in clinical trials for social phobia (social anxiety disorder), 735 subjects in clinical trials for generalized anxiety disorder and 676 subjects in clinical trials for posttraumatic stress disorder. Untoward experiences associated with this exposure were recorded by clinical investigators using descriptive terminology of their own choosing.
Consequently, it is not possible to provide a meaningful estimate of the proportion of individuals experiencing adverse experiences without first grouping similar types of untoward experiences into a limited (i.e., reduced) number of standardized experience categories.
Table 3 lists adverse experiences that occurred at an incidence of 1% or higher in short term (6-week) flexible dose (20 - 50 mg/day) placebo-controlled trials in depression. (An additional 460 patients participated in a fixed-dose placebo-controlled study.)
Table 4 enumerates adverse events that occurred at a frequency of 2% or more among patients on paroxetine who participated in placebo-controlled OCD trials of 12-weeks duration in which patients were dosed in the range of 20 - 60 mg/day and in placebo-controlled panic disorder trials of 10 - 12-weeks duration in which patients were dosed in the range of 10 - 60 mg/day, and in placebo-controlled social phobia (social anxiety disorder) trials of 12 weeks duration in which patients were dosed in a range of 20 to 50 mg/day in placebo-controlled generalized anxiety disorder trials of 8 weeks in which patients were dosed in a range from 10-50 mg/day and in placebo-controlled posttraumatic stress disorder trials of 12 weeks in which patients were dosed in a range from 20-50 mg/day.
The prescriber should be aware that these figures cannot be used to predict the incidence of side effects in the course of usual medical practice where patient characteristics and other factors differ from those which prevailed in the clinical trials. Similarly the cited incidences cannot be compared with figures obtained from other clinical investigations involving different treatments, uses and investigators. The cited frequencies do however provide the prescribing physician with some basis for estimating the relative contribution of drug and non-drug factors to the side effect incidence rate in the population studied. Reported adverse experiences were classified using a COSTART-based Dictionary terminology for the depression trials and an ADECS (a modified COSTART dictionary) for OCD and panic disorder trials. (See Tables 3, 4 and 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Male and Female Sexual Dysfunction with SSRI's:
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Male and Female Sexual Dysfunction with SSRI's: Although changes in sexual desire, sexual performance and sexual satisfaction often occur as manifestations of a psychiatric disorder, they may also be a consequence of pharmacologic treatment. In particular, some evidence suggests that selective serotonin reuptake inhibitors (SSRIs) can cause such untoward sexual experiences.
Reliable estimates of the incidence and severity of untoward experiences involving sexual desire, performance and satisfaction are difficult to obtain, however, in part because patients and physicians may be reluctant to discuss them. Accordingly, estimates of the incidence of untoward sexual experience and performance cited in product labeling are likely to underestimate their actual incidence.
In placebo-controlled clinical trials involving more than 3,200 patients, the ranges for the reported incidence of sexual side effects in males and females with major depressive disorder, OCD, panic disorder, social anxiety disorder, GAD and PTSD are displayed in Table 6 as follows. (See Table 6.)
Click on icon to see table/diagram/image
There are no adequate and well-controlled studies examining sexual dysfunction with paroxetine treatment.
Paroxetine treatment has been associated with several cases of priapism. In those cases with a known outcome, patients recovered without sequelae.
While it is difficult to know the precise risk of sexual dysfunction associated with the use of SSRIs, physicians should routinely inquire about such possible side effects.
In the tabulations which follow, a COSTART or modified COSTART-based Dictionary terminology has been used to classify reported adverse experiences. The frequencies presented therefore represent the portion of the 4126, 542, 469, 522, 735 and 676 paroxetine-exposed individuals in depression, OCD, panic, social phobia (social anxiety disorder), generalized anxiety disorder and post-traumatic stress disorder trials, respectively, who experienced an event of the type cited on at least one occasion while receiving paroxetine. Experiences are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent experiences are defined as those occurring on one or more occasion in at least 1/100 patients; infrequent adverse experiences are those occurring in less than 1/100 but at least 1/1000 patients; rare experiences are those occurring in less than 1/1000 patients.
All adverse experiences are included except those already listed in Table 3 and Table 4, those reported in terms so general as to be uninformative and those experiences for which the drug cause was remote. It is important to emphasize that although the experiences reported did occur during treatment with paroxetine, they were not necessarily caused by it.
Body as a Whole: Frequent: Malaise, pain. Infrequent: Allergic reaction, chills, face edema, infection, moniliasis, neck pain, overdose. Rare: Abnormal laboratory value, abscess, adrenergic syndrome, cellulitis, chills and fever, cyst, hernia, intentional overdose, neck rigidity, pelvic pain, peritonitis, substernal chest pain, sepsis, ulcer.
Cardiovascular System: Frequent: Hypertension, syncope, tachycardia. Infrequent: Bradycardia, conduction abnormalities, electrocardiogram abnormal, hypotension, migraine, ventricular extrasystoles.
Rare: Angina pectoris, arrhythmia, atrial arrhythmia, atrial fibrillation, bundle branch block, cardiac disorder, cerebral ischemia, cerebrovascular accident, cerebrovascular disorder, congestive heart failure, extrasystoles, low cardiac output, myocardial infarct, myocardial ischemia, pallor, phlebitis, pulmonary embolus, supraventricular extrasystoles, thrombosis, varicose vein, vascular disorder, vascular headache.
Dermatological: Frequent: Pruritus. Infrequent: Acne, alopecia, dry skin, ecchymosis, eczema, furunculosis, herpes simplex, urticaria. Rare: Angioedema, contact dermatitis, erythema nodosum, exfoliative dermatitis, herpes zoster, maculopapular rash, photosensitivity, skin discolouration, skin ulcer, skin hypertrophy, sweating decreased.
Endocrine: Rare: Diabetes mellitus, fertility decreased female, goiter, hyperthyroidism, hypothyroidism, thyroiditis.
Gastrointestinal: Frequent: Nausea and vomiting. Infrequent: Bruxism, buccal cavity disorders, dysphagia, eructation, gastroenteritis, gastrointestinal flu, glossitis, increased salivation, liver function tests abnormal, mouth ulceration, vomiting and diarrhea, rectal hemorrhage. Rare: Aphthous stomatitis, bloody diarrhea, bulimia, cardiospasm, colitis, duodenitis, esophagitis, fecal impaction, fecal incontinence, gastritis, gingivitis, hematemesis, hepatitis, ileitis, ileus, jaundice, melena, peptic ulcer, salivary gland enlargement, sialadenitis, stomach ulcer, stomatitis, tongue edema, tooth caries.
Hematologic and Lymphatic: Infrequent: Anemia, leukopenia, lymphadenopathy, purpura, WBC abnormality. Rare: Abnormal bleeding, predominantly of the skin and mucous membranes (mostly ecchymosis), bleeding increased, eosinophilia, iron deficiency anemia, leukocytosis, lymphedema, lymphocytosis, microcytic anemia, monocytosis, normocytic anemia, thrombocytopenia.
Metabolic and Nutritional: Frequent: Weight gain, weight loss. Infrequent: Edema, hyperglycemia, peripheral edema, thirst. Rare: Alkaline phosphatase increased, bilirubinemia, cachexia, dehydration, gout, hypercholesteremia, hypocalcemia, hypoglycemia, hypokalemia, hyponatremia (predominantly in the elderly) which is sometimes due to syndrome of inappropriate anti-diuretic hormone secretion (SIADH), non-protein nitrogen (NPN) increased, obesity, SGOT increased, SGPT increased.
Musculoskeletal: Infrequent:
Arthralgia, arthritis, traumatic fracture. Rare: Arthrosis, bone disorder, bursitis, cartilage disorder, myositis, osteoporosis, tetany.
Nervous System: Frequent: CNS stimulation, concentration impaired, depression, emotional lability, vertigo. Infrequent: Akinesia, alcohol abuse, amnesia, ataxia, convulsion, depersonalization, hallucinations, hyperkinesia, hypertonia, incoordination, lack of emotion, manic reaction, paranoid reaction, thinking abnormal, hypesthesia. Rare: Abnormal electroencephalogram, abnormal gait, antisocial reaction, brain edema, choreoathetosis, circumoral paresthesia, confusion, delirium, delusions, diplopia, drug dependence, dysarthria, dyskinesia, dystonia, euphoria, fasciculations, grand mal convulsion, hostility, hyperalgesia, hypokinesia, hysteria, libido increased, manic depressive reaction, meningitis, myelitis, neuralgia, neuropathy, nystagmus, psychosis, psychotic depression, reflexes increased, stupor, torticollis, withdrawal syndrome.
Respiratory System: Frequent: Cough increased, rhinitis. Infrequent: Asthma, bronchitis, dyspnea, epistaxis, hyperventilation, pneumonia, respiratory flu, sinusitis. Rare: Hiccup, lung fibrosis, sputum increased, stridor, trachea disorder, voice alteration.
Special Senses: Infrequent: Abnormality of accommodation, conjunctivitis, ear pain, eye pain, mydriasis, otitis media, tinnitus. Rare: Amblyopia, cataract specified, conjunctival edema, corneal lesion, corneal ulcer, exophthalmos, eye hemorrhage, acute glaucoma, hyperacusis, otitis externa, photophobia, retinal hemorrhage, taste loss, anisocoria, deafness, keratoconjunctivitis.
Urogenital System: Infrequent: Abortion*, amenorrhea*, breast pain*, cystitis, dysmenorrhea*, dysuria, menorrhagia*, nocturia, polyuria, urinary incontinence, urinary retention, urinary tract infection, urinary urgency, vaginitis*. Rare: Breast atrophy*, cervix disorder*, endometrial disorder*, female lactation*, hematuria, kidney calculus, kidney function abnormal, kidney pain, mastitis*, nephritis, oliguria, salpingitis*, spermatogenesis arrest*, urethritis, urinary casts, urine abnormality, uterine neoplasm*, vaginal moniliasis*.
* Incidence corrected for gender.
Post-marketing Reports: Adverse events not listed as previously mentioned which have been reported since market introduction in patients taking paroxetine include acute pancreatitis, hepatic events such as elevation of hepatic enzymes, and hepatitis, sometimes associated with jaundice, and/or liver failure (in very rare circumstances, with fatal outcomes), Guillain-Barré syndrome, toxic epidermal necrolysis, priapism, thrombocytopenia, aggravated hypertension, syndrome of inappropriate ADH secretion, symptoms suggestive of hyperprolactinemia and galactorrhea, blurred vision, neuroleptic malignant syndrome-like events; extrapyramidal symptoms which have included akathisia, bradykinesia, cogwheel rigidity, dystonia, hypertonia, oculogyric crisis which has been associated with concomitant use of pimozide, tremor and trismus; and serotonin syndrome, associated in some cases with concomitant use of serotonergic drugs and with drugs which may have impaired paroxetine metabolism (symptoms have included agitation, confusion, diaphoresis, hallucinations, hyperreflexia, myoclonus, shivering, tachycardia and tremor). There has been a case report of an elevated phenytoin level after 4 weeks of paroxetine and phenytoin co-administration. There has been a case report of severe hypotension when paroxetine was added to chronic metoprolol treatment. The causal relationship between paroxetine and the emergence of these events has not been established.
There have been spontaneous reports of adverse events upon the discontinuation of paroxetine (particularly when abrupt), including but not limited to the following: dizziness, sensory disturbances (including paresthesias and electric shock sensations), agitation/restlessness, anxiety, nausea, vomiting, sweating, headache, and sleep disturbance. These events are generally self-limiting. Symptoms associated with discontinuation have also been reported for other selective serotonin reuptake inhibitors (see PRECAUTIONS).



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image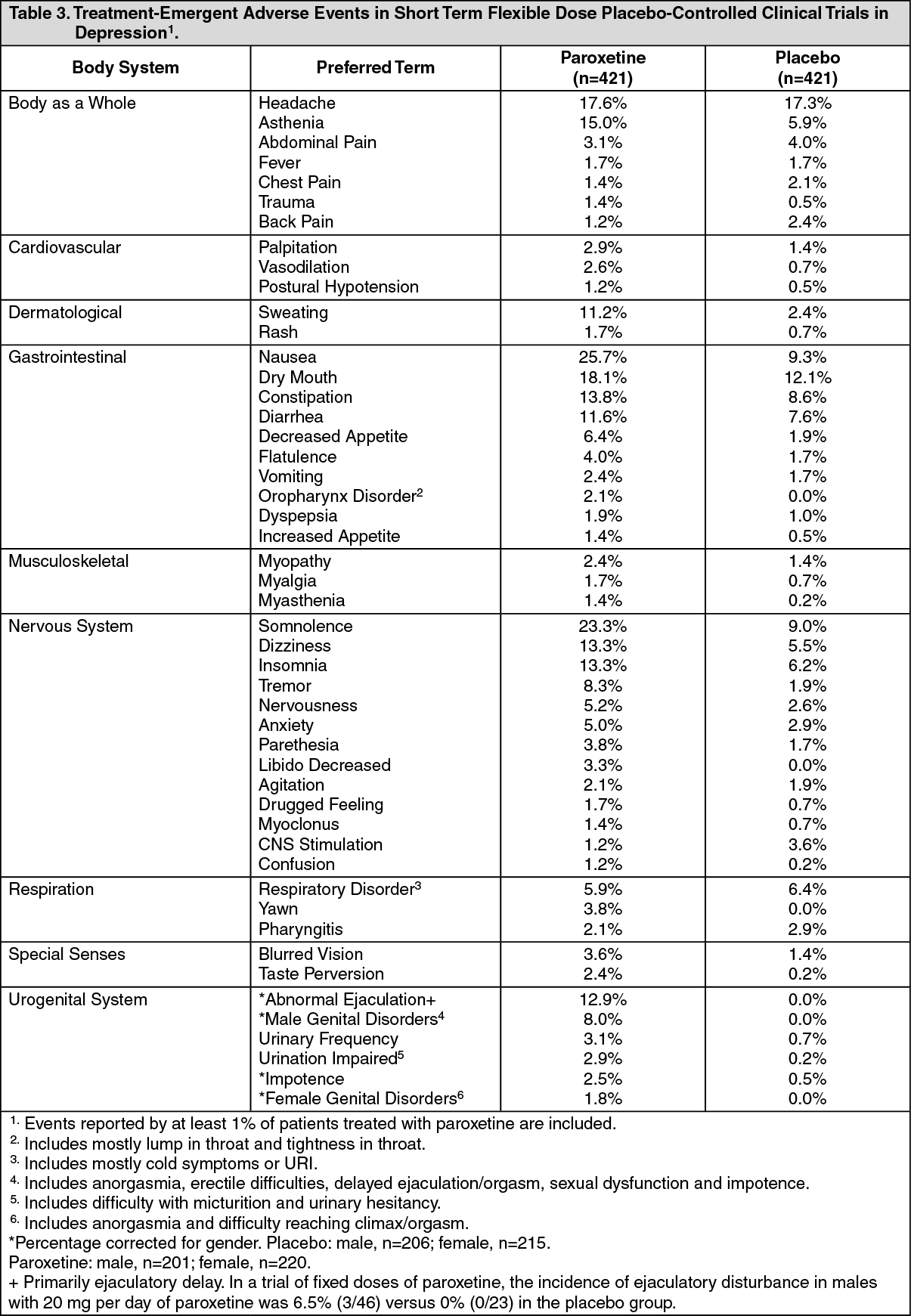 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image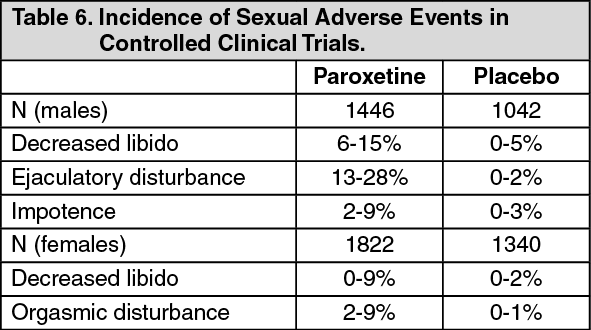 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out