Pharmacotherapeutic group: Other antineoplastic agents, monoclonal antibodies.
ATC code: L01FF04.
Pharmacology: Pharmacodynamics: Mechanism of action: Avelumab is a human immunoglobulin G1 (IgG1) monoclonal antibody directed against programmed death ligand 1 (PD-L1). Avelumab binds PD-L1 and blocks the interaction between PD-L1 and the programmed death 1 (PD-1) and B7.1 receptors. This removes the suppressive effects of PD-L1 on cytotoxic CD8+ T-cells, resulting in the restoration of anti-tumour T-cell responses.
Avelumab has also shown to induce natural killer (NK) cell-mediated direct tumour cell lysis via antibody-dependent cell-mediated cytotoxicity (ADCC).
Clinical efficacy and safety: Merkel cell carcinoma (study EMR100070-003): The efficacy and safety of avelumab was investigated in the study EMR100070-003 with two parts. Part A was a single-arm, multi-centre study conducted in patients with histologically confirmed metastatic MCC, whose disease had progressed on or after chemotherapy administered for distant metastatic disease, with a life expectancy of more than 3 months. Part B included patients with histologically confirmed metastatic MCC who were treatment-naïve to systemic therapy in the metastatic setting.
Patients with active or a history of central nervous system (CNS) metastasis; active or a history of autoimmune disease; a history of other malignancies within the last 5 years; organ transplant; conditions requiring therapeutic immune suppression or active infection with HIV, or hepatitis B or C were excluded.
Patients received avelumab at a dose of 10 mg/kg every 2 weeks until disease progression or unacceptable toxicity. Patients with radiological disease progression not associated with significant clinical deterioration, defined as no new or worsening symptoms, no change in performance status for greater than two weeks, and no need for salvage therapy could continue treatment.
Tumour response assessments were performed every 6 weeks, as assessed by an Independent Endpoint Review Committee (IERC) using Response Evaluation Criteria in Solid Tumours (RECIST) v1.1.
For Part A, the major efficacy outcome measure was confirmed best overall response (BOR); secondary efficacy outcome measures included duration of response (DOR), and progression-free survival (PFS).
For Part A, the efficacy analysis was conducted in all 88 patients after a minimum follow-up of 18 months Patients received a median of 7 doses of avelumab (range: 1 dose to 61 doses), and the median duration of treatment was 17 weeks (range: 2 weeks to 132 weeks).
Of the 88 patients, 65 (74%) were male, the median age was 73 years (range 33 years to 88 years), 81 (92%) patients were Caucasian, and 49 (56%) patients and 39 (44%) patients with an Eastern Cooperative Oncology Group (ECOG) performance status 0 and 1, respectively.
Overall, 52 (59%) patients were reported to have had 1 prior anti-cancer therapy for MCC, 26 (30%) with 2 prior therapies, and 10 (11%) with 3 or more prior therapies. Forty-seven (53%) of the patients had visceral metastases.
Table 1 summarises efficacy endpoints in patients receiving avelumab at the recommended dose for study EMR100070-003, Part A. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
The median time to response was 6 weeks (range: 6 weeks to 36 weeks) after the first dose of avelumab. Twenty-two out of 29 (76%) patients with response were reported to have responded within 7 weeks after the first dose of avelumab.
The Kaplan-Meier estimates of PFS of the 88 patients (Part A) with metastatic MCC is presented in Figure 1. (See Figure 1.)
Click on icon to see table/diagram/image
Tumour samples were evaluated for PD-L1 tumour cell expression, and for Merkel cell polyomavirus (MCV) using an investigational immunohistochemistry (IHC) assay. Table 2 summarises the PD-L1 expression and MCV status of patients with metastatic MCC in study EMR100070-003 (Part A). (See Table 2.)
Click on icon to see table/diagram/image
The clinical utility of PD-L1 as a predictive biomarker in MCC has not been established.
For Part B, the major efficacy outcome measure was durable response, defined as objective response (complete response (CR) or partial response (PR)) with a duration of at least 6 months; secondary outcome measures included BOR, DOR, PFS, and OS.
For Part B, an interim analysis of efficacy was conducted with 39 patients who received at least one dose. Of those, 30 (77%) were males, the median age was 75 years (range: 47 years to 88 years), 33 (85%) patients were Caucasian, and 31 (79%) patients and 8 (21%) patients had an ECOG performance status 0 and 1, respectively. Twenty-nine patients had at least 13 weeks of follow-up at the time of the data cut-off.
Table 3 summarises efficacy endpoints in patients receiving avelumab at the recommended dose for study EMR100070-003, Part B. (See Table 3.)
Click on icon to see table/diagram/image
Figure 2 presents the Kaplan-Meier estimates for PFS for the 39 patients enrolled into Part B who received at least one dose of study drug prior to the data cut-off for the interim analysis. (See Figure 2.)
Click on icon to see table/diagram/image
Locally advanced or metastatic urothelial carcinoma (study B9991001): The efficacy and safety of avelumab was demonstrated in study B9991001, a randomised, multi-center, open-label study conducted in 700 patients with unresectable, locally advanced or metastatic urothelial carcinoma whose disease had not progressed with first-line platinum-based induction chemotherapy. Patients with autoimmune disease or a medical condition that required immunosuppression were excluded. Randomization was stratified by best response to chemotherapy (CR/PR vs. stable disease [SD]) and site of metastasis (visceral vs. non-visceral) at the time of initiating first-line induction chemotherapy. Patients were randomised (1:1) to receive either avelumab 10 mg/kg intravenous infusion every 2 weeks plus best supportive care (BSC) or BSC alone.
Treatment with avelumab continued until Response Evaluation Criteria in Solid Tumours (RECIST) v1.1-defined progression of disease by Blinded Independent Central Review (BICR) assessment or unacceptable toxicity. Administration of avelumab was permitted beyond RECIST-defined disease progression if the patient was clinically stable and considered to be deriving clinical benefit by the investigator. Assessment of tumour status was performed at baseline, 8 weeks after randomization, then every 8 weeks up to 12 months after randomization, and every 12 weeks thereafter until documented confirmed disease progression based on BICR assessment per RECIST v1.1. Demographic and baseline characteristics were generally well balanced between the avelumab plus BSC and the BSC alone arm. Baseline characteristics were a median age of 69 years (range: 32 to 90), 66% of patients were 65 years or older, 77% were male, 67% were White, and the ECOG PS was 0 (61%) or 1 (39%) for both arms.
For first-line induction chemotherapy, 56% of patients received cisplatin plus gemcitabine, 38% of patients received carboplatin plus gemcitabine and 6% of patients received cisplatin plus gemcitabine and carboplatin plus gemcitabine (i.e. these patients received one or more cycles of each combination). Best response to first-line induction chemotherapy was CR or PR (72%) or SD (28%). Sites of metastasis prior to chemotherapy were visceral (55%) or non-visceral (45%). Fifty-one percent of patients had PD-L1-positive tumours. Six percent of patients in the avelumab plus BSC arm and 44% of patients in the BSC alone arm received another PD-1/PD-L1 checkpoint inhibitor after discontinuation of treatment.
The primary efficacy outcome measure was overall survival (OS) in all randomized patients and in patients with PD-L1-positive tumours. Progression-free survival (PFS) based on BICR assessment per RECIST v1.1 was an additional efficacy outcome measure. Efficacy outcomes were measured from time of randomisation after 4 to 6 cycles of platinum-based induction chemotherapy.
Efficacy results are presented as follows. (See Table 4, Figures 3 and 4.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In addition, a statistically significant improvement in OS was demonstrated in patients with PD-L1-positive tumours for avelumab plus BSC compared to BSC alone (HR 0.56; 95% CI: 0.40, 0.79; 2-sided p-value 0.0007). The median OS was not reached (95% CI: 20.3 months, not reached) in the avelumab plus BSC arm, and 17.1 months (95% CI: 13.5, 23.7) in the BSC alone arm. In an exploratory analysis of patients with PD-L1-negative tumours (n=271, 39%), the OS hazard ratio was 0.85 (95% CI: 0.62, 1.18). (See Figure 5.)
Click on icon to see table/diagram/image
Consistent results were observed across pre-specified subgroups, including best response to first-line induction chemotherapy, sites of metastasis, as shown in Figure 6. (See Figure 6.)
Click on icon to see table/diagram/image
Patient reported outcomes (PRO): Patient reported outcomes of physical and emotional disease related symptoms, treatment side effects, and function and well-being were collected using the FACT/NCCN Bladder Symptom Index (FBlSI-18). No detrimental effects were observed when adding avelumab maintenance therapy to BSC compared to BSC alone as measured by FBlSI-18 during treatment period.
Renal cell carcinoma (study B9991003): The efficacy and safety of avelumab in combination with axitinib was demonstrated in study B9991003, a randomised, multicentre, open-label study of avelumab in combination with axitinib in 886 patients with untreated advanced or metastatic RCC with a clear-cell component.
Patients were included irrespective of prognostic risk groups or tumour PD-L1 expression and had to have at least one measurable lesion as defined by Response Evaluation Criteria in Solid Tumours (RECIST) version 1.1 that was not been previously irradiated. Patients with prior systemic therapy directed at advanced or metastatic RCC; prior systemic immunotherapy treatment with IL-2, IFN-α, anti-PD-1, anti-PD-L1, or anti-CTLA-4 antibodies, or active brain metastasis; active autoimmune disease that might deteriorate when receiving an immunostimulatory agents; a history of other malignancies within the last 5 years; organ transplant were ineligible.
Randomization was stratified according to Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) (0 vs. 1) and region (United States vs. Canada/Western Europe vs. the rest of the world). Patients were randomised (1:1) to one of the following treatment arms: Avelumab 10 mg/kg intravenous infusion every 2 weeks in combination with axitinib 5 mg twice daily orally (N=442). Patients who tolerated axitinib 5 mg twice daily without Grade 2 or greater axitinib-related adverse events for 2 consecutive weeks could increase to 7 mg and then subsequently to 10 mg twice daily. Axitinib could be interrupted or reduced to 3 mg twice daily and subsequently to 2 mg twice daily to manage toxicity.
Sunitinib 50 mg once daily orally for 4 weeks followed by 2 weeks off (N=444) until radiographic or clinical progression or unacceptable toxicity.
Treatment with avelumab and axitinib continued until RECIST v1.1-defined progression of disease by Blinded Independent Central Review (BICR) assessment or unacceptable toxicity. Administration of avelumab and axitinib was permitted beyond RECIST-defined disease progression based on investigator's assessment of the patient's benefit-risk and clinical condition, including performance status, clinical symptoms, adverse events and laboratory data. The majority (n=160, 71.4%) of the patients with progressive disease continued treatment with both medicinal products after progression. Assessment of tumour status was performed at baseline, after randomisation at 6 weeks, then every 6 weeks thereafter up to 18 months after randomisation, and every 12 weeks thereafter until documented confirmed disease progression by BICR.
The primary efficacy endpoints were progression-free survival (PFS), as assessed by BICR using RECIST v1.1 and overall survival (OS) in the first-line treatment of patients with advanced RCC who have PD-L1-positive tumours (PD-L1 expression level ≥1%). The key secondary endpoints were PFS based on BICR assessment per RECIST v1.1 and OS irrespective of PD-L1 expression. PD-L1 status was determined by immunohistochemistry. Additional secondary endpoints included objective response (OR), time to response (TTR) and duration of response (DOR).
Study population characteristics were: median age of 61 years (range: 27.0 to 88.0), 38% of patients were 65 years or older, 75% were male, 75% were White, and the ECOG performance score was 0 (63%) or 1 (37%).
Patient distribution by International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) risk groups was 21% favourable, 62% intermediate, and 16% poor. Patient distribution by Memorial Sloan-Kettering Cancer Center (MSKCC) risk groups was 22% favourable, 65% intermediate, and 11% poor.
Efficacy results are presented in Table 4 and Figure 3 based on a data cut-off date of 28 January 2019.
With a median OS follow-up of 19 months, OS data were immature with 27% deaths. The observed hazard ratio (HR) for OS was 0.80 (95% CI: 0.616, 1.027) for avelumab in combination with axitinib compared to sunitinib. (See Table 5 and Figure 7.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Improvement of PFS was observed across pre-specified subgroups. (See Figure 8.)
Click on icon to see table/diagram/image
Pharmacokinetics: Avelumab pharmacokinetics (PK) was assessed using a population PK approach for avelumab as monotherapy and avelumab in combination with axitinib.
Based on a population PK analysis for avelumab as monotherapy and in combination with axitinib, there are no expected clinically meaningful differences in exposure of avelumab between settings administered every 2 weeks at 800 mg or 10 mg/kg.
Distribution: Avelumab is expected to be distributed in the systemic circulation and to a lesser extent in the extracellular space. The volume of distribution at steady state was 4.72 L.
Consistent with a limited extravascular distribution, the volume of distribution of avelumab at steady state is small. As expected for an antibody, avelumab does not bind to plasma proteins in a specific manner.
Elimination: Based on a population pharmacokinetic analysis from 1,629 patients, the value of total systemic clearance (CL) is 0.59 L/day. In the supplemental analysis, avelumab CL was found to decrease over time: the largest mean maximal reduction (% coefficient of variation [CV%]) from baseline value with different tumour types was approximately 32.1% (CV 36.2%).
Steady-state concentrations of avelumab were reached after approximately 4 to 6 weeks (2 to 3 cycles) of repeated dosing at 10 mg/kg every 2 weeks, and systemic accumulation was approximately 1.25-fold.
The elimination half-life (t
½) at the recommended dose is 6.1 days based on the population PK analysis.
Linearity/non-linearity: The exposure of avelumab increased dose-proportionally in the dose range of 10 mg/kg to 20 mg/kg every 2 weeks.
When avelumab 10 mg/kg was administered in combination with axitinib 5 mg, the respective exposures of avelumab and axitinib were unchanged compared to the single agents. There was no evidence to suggest a clinically relevant change of avelumab clearance over time in patients with advanced RCC.
Special populations: A population pharmacokinetic analysis suggested no difference in the total systemic clearance of avelumab based on age, gender, race, PD-L1 status, tumour burden, renal impairment and mild or moderate hepatic impairment.
Total systemic clearance increases with body weight. Steady-state exposure was approximately uniform over a wide range of body weights (30 to 204 kg) for body weight normalised dosing.
Renal impairment: No clinically important differences in the clearance of avelumab were found between patients with mild (glomerular filtration rate (GFR) 60 to 89 mL/min, Cockcroft-Gault Creatinine Clearance (CrCL); n=623), moderate (GFR 30 to 59 mL/min, n=320) and patients with normal (GFR ≥90 mL/min, n=671) renal function.
Avelumab has not been studied in patients with severe renal impairment (GFR 15 to 29 mL/min).
Hepatic impairment: No clinically important differences in the clearance of avelumab were found between patients with mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin between 1 and 1.5 times ULN, n=217) and normal hepatic function (bilirubin and AST ≤ ULN, n=1,388) in a population PK analysis. Hepatic impairment was defined by National Cancer Institute (NCI) criteria of hepatic dysfunction.
Avelumab has not been studied in patients with moderate hepatic impairment (bilirubin between 1.5 and 3 times ULN) or severe hepatic impairment (bilirubin > 3 times ULN).
Toxicology: Preclinical safety data: Non-clinical data reveal no special hazard for humans based on conventional studies of repeated dose toxicity in Cynomolgus monkeys administered intravenously doses of 20, 60 or 140 mg/kg once a week for 1 month and 3 months, followed by a 2-month recovery period after the 3-month dosing period. Perivascular mononuclear cell cuffing was observed in the brain and spinal cord of monkeys treated with avelumab at ≥20 mg/kg for 3 months. Although there was no clear dose-response relationship, it cannot be excluded that this finding was related to avelumab treatment.
Animal reproduction studies have not been conducted with avelumab. The PD-1/PD-L1 pathway is thought to be involved in maintaining tolerance to the foetus throughout pregnancy. Blockade of PD-L1 signalling has been shown in murine models of pregnancy to disrupt tolerance to the foetus and to result in an increase in foetal loss. These results indicate a potential risk that administration of avelumab during pregnancy could cause foetal harm, including increased rates of abortion or stillbirth.
No studies have been conducted to assess the potential of avelumab for carcinogenicity or genotoxicity.
Fertility studies have not been conducted with avelumab. In 1-month and 3-month repeat-dose toxicology studies in monkeys, there were no notable effects in the female reproductive organs. Many of the male monkeys used in these studies were sexually immature and thus no explicit conclusions regarding effects on male reproductive organs can be made.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image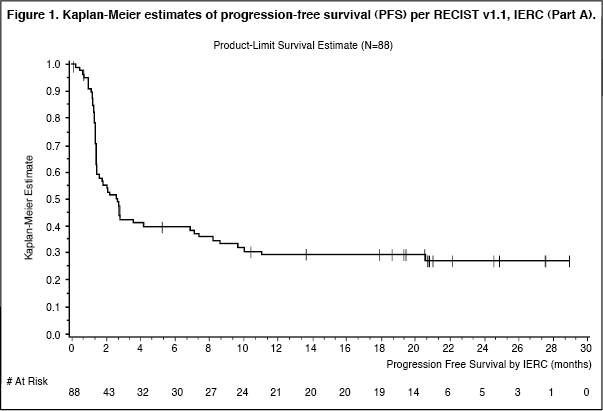 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image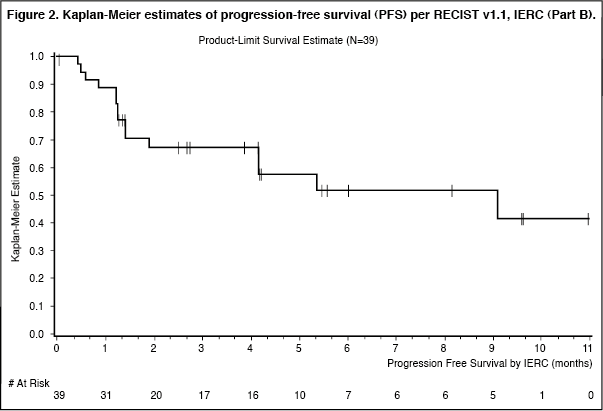 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image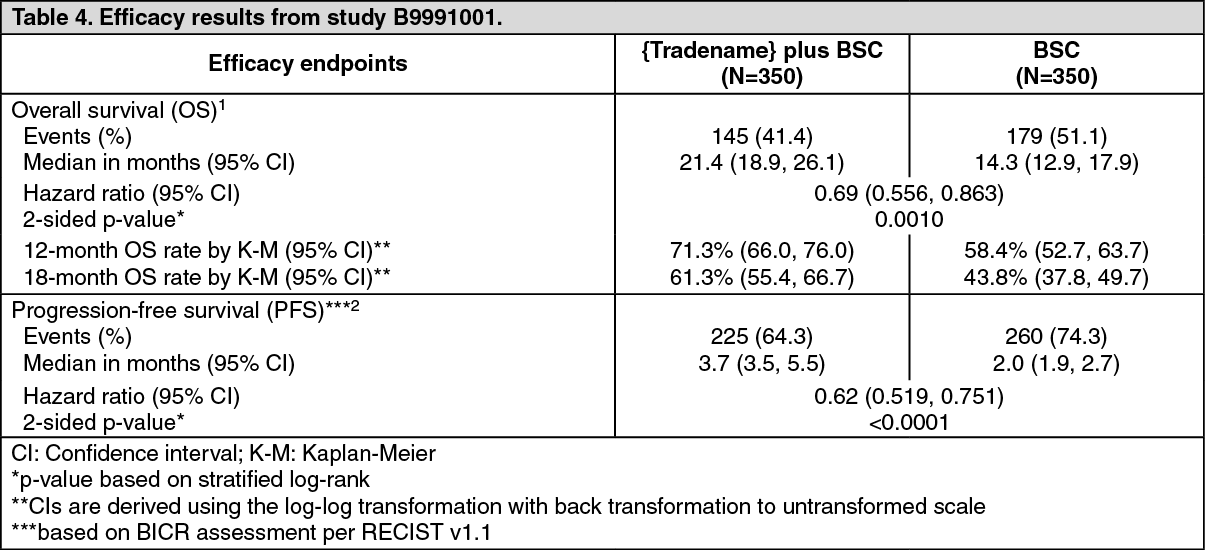 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image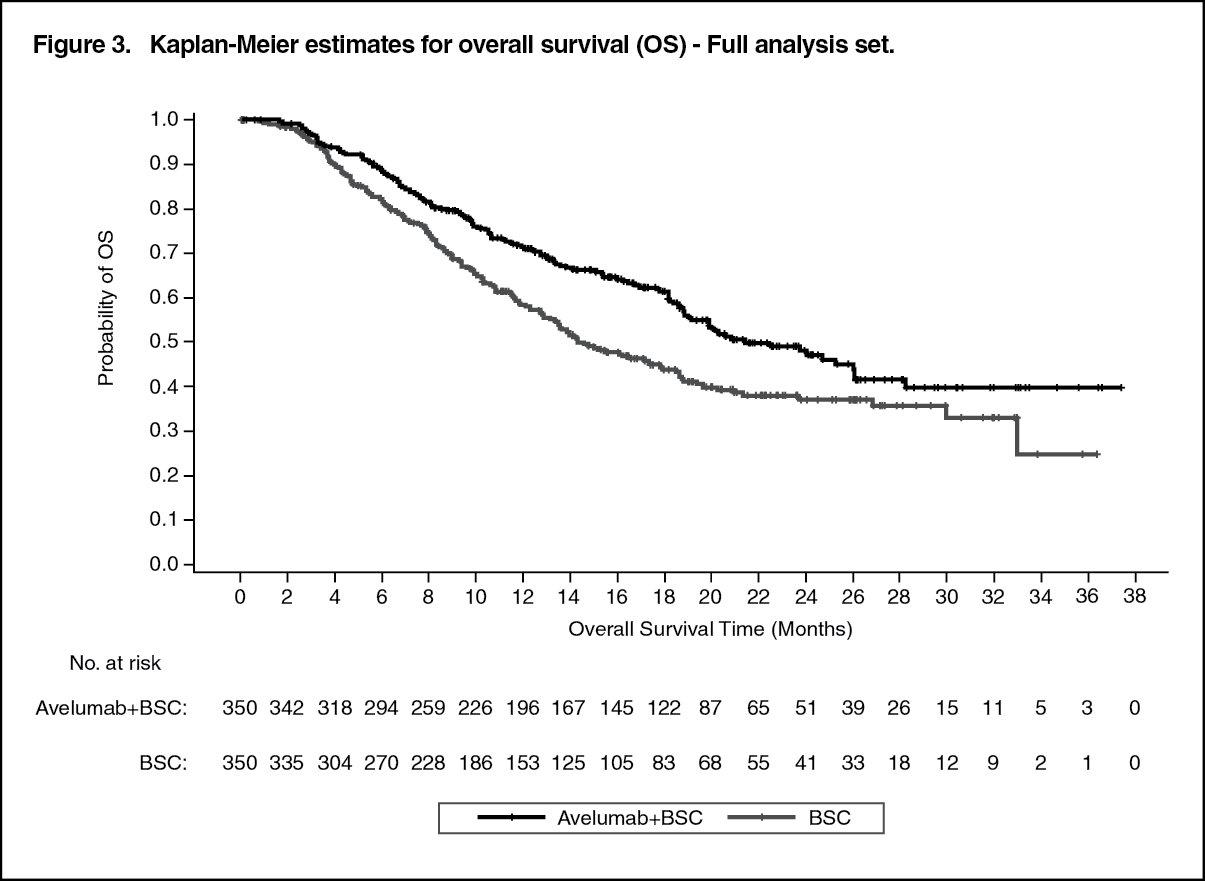 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image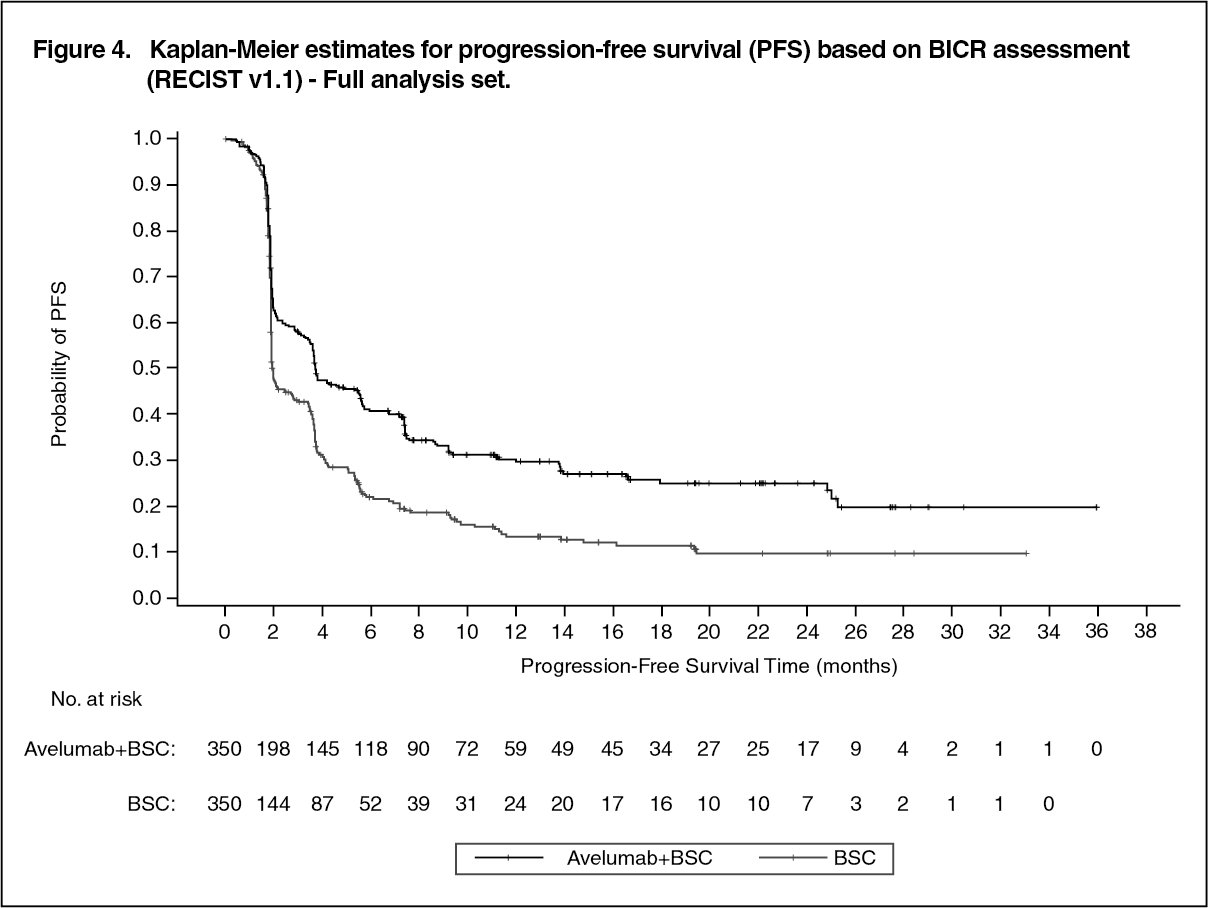 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image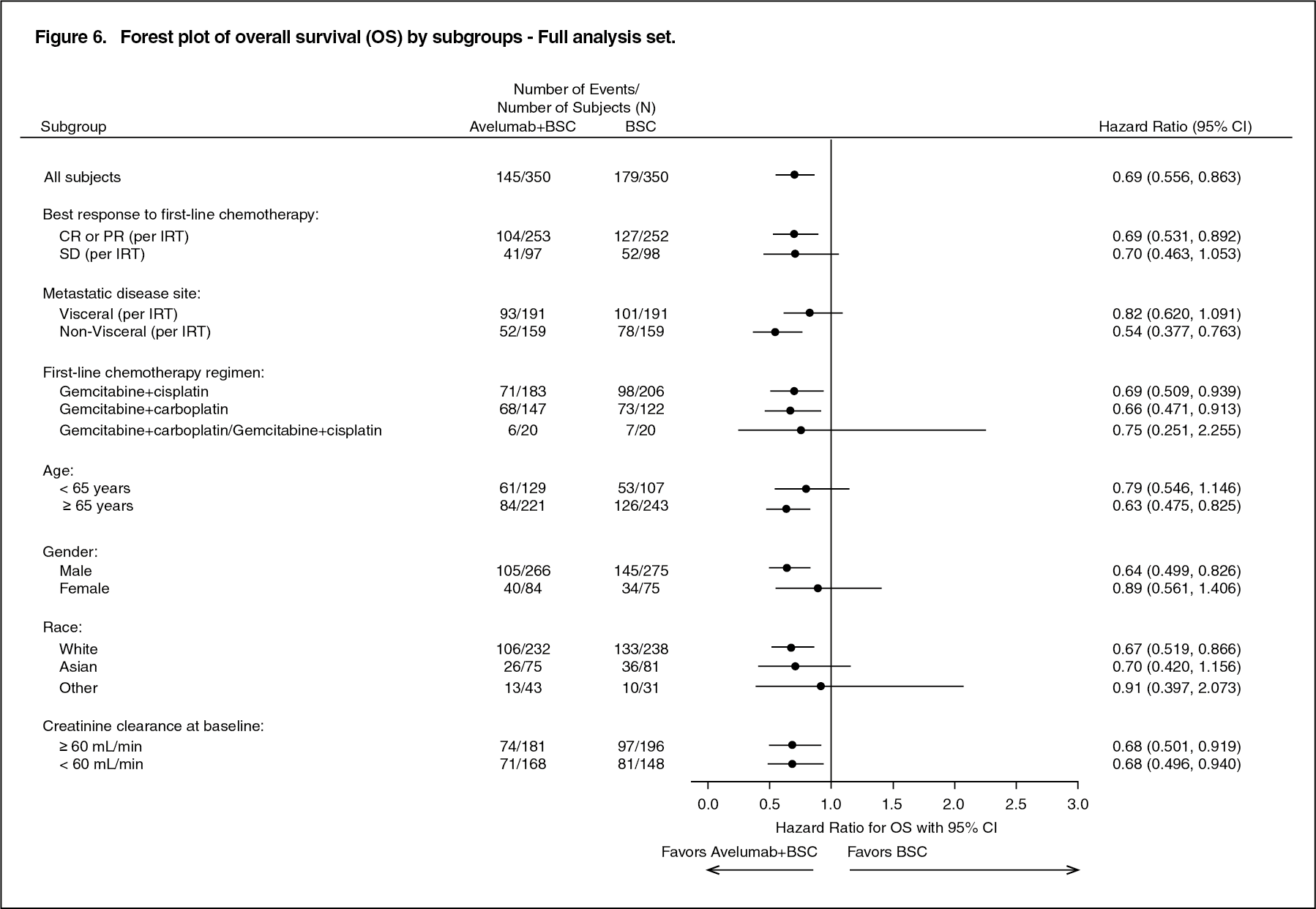 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image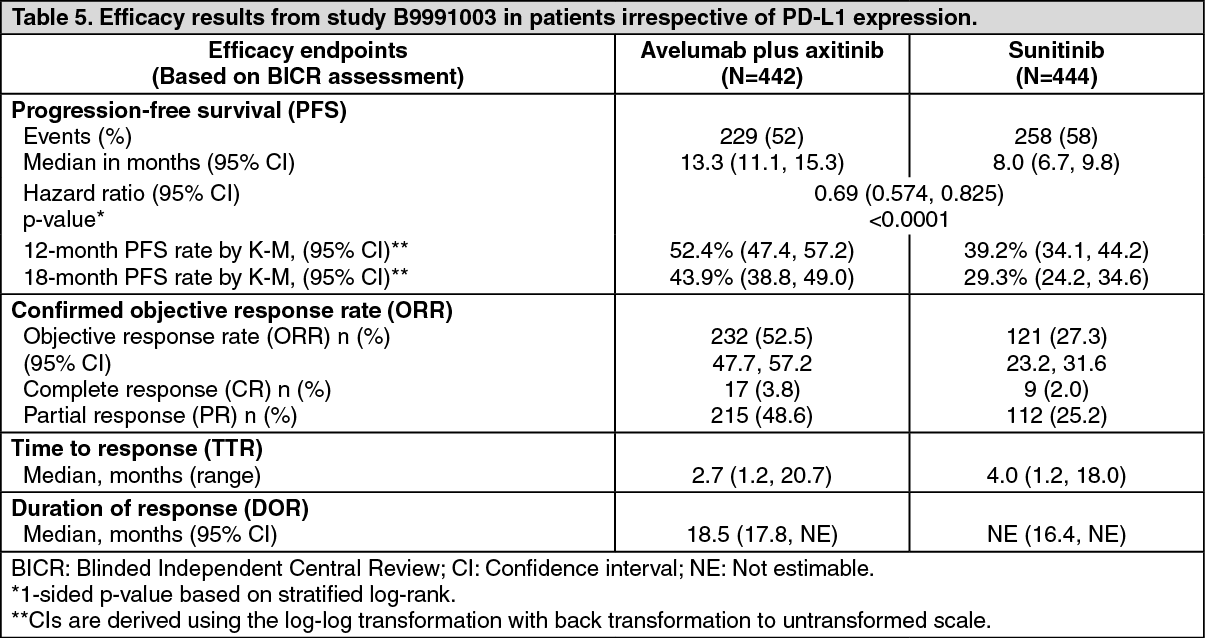 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image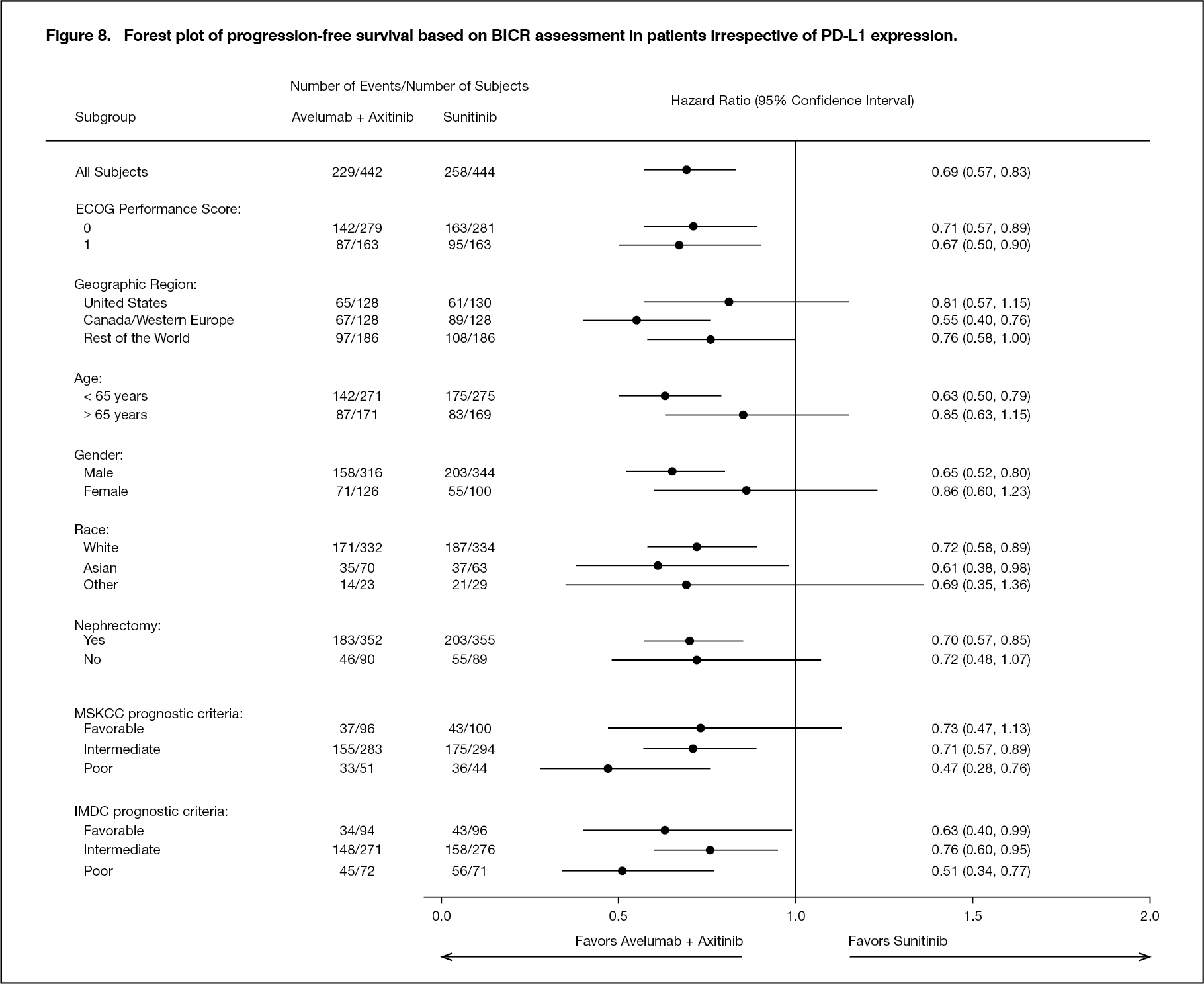 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out