


 Sign Out
Sign Out


Pharmacology: Pharmacodynamics: Mechanism of Action: Degarelix is a selective gonadotrophin releasing hormone (GnRH) antagonist that competitively and reversibly binds to the pituitary GnRH receptors, thereby rapidly reducing the release of the gonadotrophins, luteinizing hormone (LH) and follicle stimulating hormone (FSH), and thereby reducing the secretion of testosterone (T) by the testes. Prostatic carcinoma is known to be androgen sensitive and responds to treatment that removes the source of androgen. Unlike GnRH agonists, GnRH antagonists do not induce a LH surge with subsequent testosterone surge/tumour stimulation and potential symptomatic flare after the initiation of treatment.
A single dose of 240 mg degarelix, followed by a monthly maintenance dose of 80 mg, rapidly causes a decrease in the concentrations of LH, FSH and subsequently testosterone. The plasma concentration of dihydrotestosterone (DHT) decreases in a similar manner to testosterone.
Degarelix is effective in achieving and maintaining testosterone suppression well below medical castration level of 0.5 ng/ml. Maintenance monthly dosing of 80 mg resulted in sustained testosterone suppression in 97% of patients for at least one year. No testosterone microsurges were observed after re-injection during degarelix treatment. Median testosterone levels after one year of treatment were 0.087 ng/ml (interquartile range 0.06-0.15) N=167.
Results of the confirmatory Phase III study: The efficacy and safety of degarelix was evaluated in an open-label, multi-centre, randomised, active comparator controlled, parallel-group study. The study investigated the efficacy and safety of two different degarelix monthly dosing regimens with a starting dose of 240 mg (40 mg/ml) followed by monthly doses subcutaneous administration of 160 mg (40 mg/ml) or 80 mg (20 mg/ml), in comparison to monthly intramuscular administration of 7.5 mg leuprorelin in patients with prostate cancer requiring androgen deprivation therapy. In total 620 patients were randomised to one of the three treatment groups, of which 504 (81%) patients completed the study. In the degarelix 240/80 mg treatment group 41 (20%) patients discontinued the study, as compared to 32 (16%) patients in the leuprorelin group.
Of the 610 patients treated: 31% had localised prostate cancer, 29% had locally advanced prostate cancer, 20% had metastatic prostate cancer, 7% had an unknown metastatic status, 13% had previous curative intent surgery or radiation and a rising PSA.
Baseline demographics were similar between the arms. The median age was 74 years (range 47 to 98 years). The primary objective was to demonstrate that degarelix is effective with respect to achieving and maintaining testosterone suppression to below 0.5 ng/ml, during 12 months of treatment.
The lowest effective maintenance dose of 80 mg degarelix was chosen.
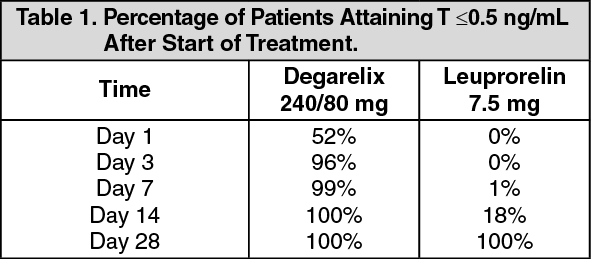
Attainment of serum testosterone (T) ≤0.5 ng/ml: FIRMAGON is effective in achieving fast testosterone suppression, (see Table 1).
 Click on icon to see table/diagram/image
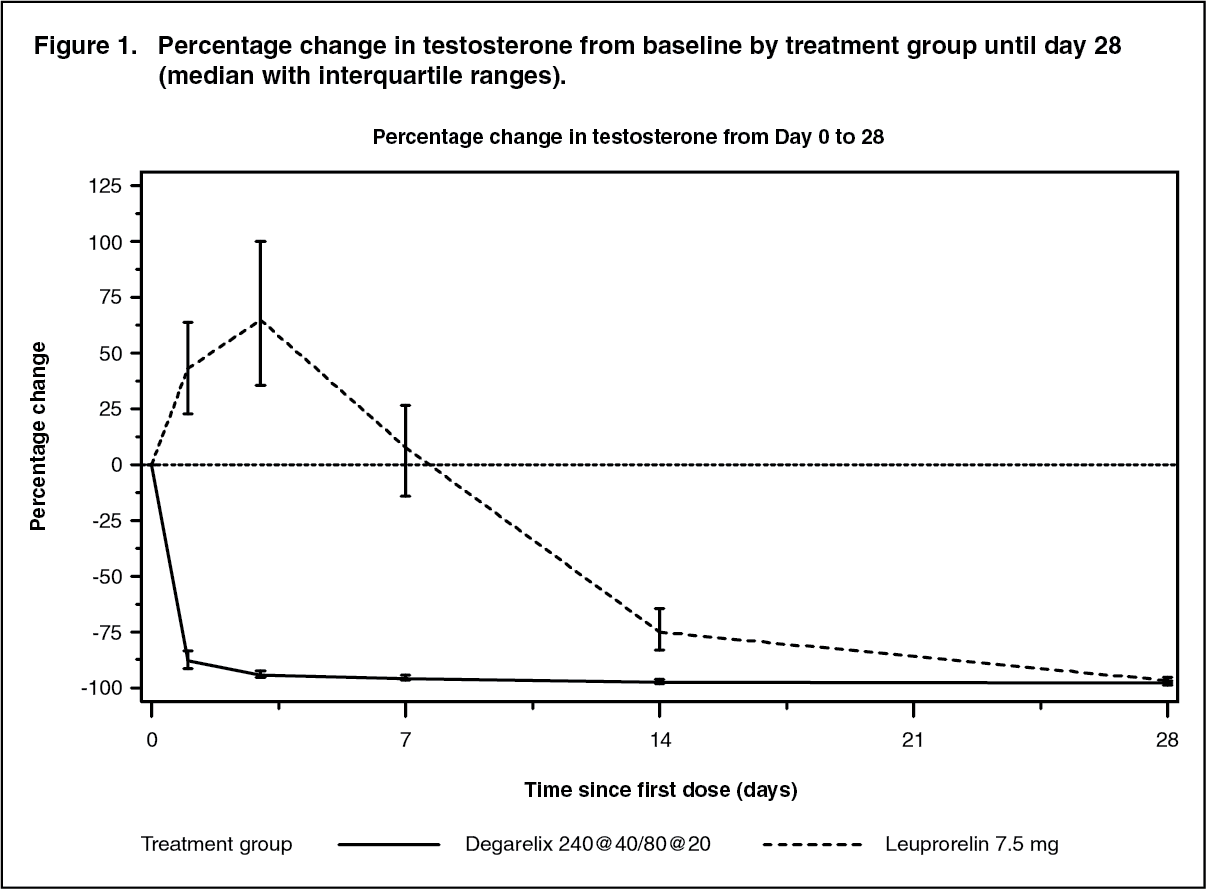
Click on icon to see table/diagram/imageAvoidance of testosterone surge: Surge was defined as testosterone exceeding baseline by ≥15% within first 2 weeks. None of the degarelix-treated patients experienced a testosterone surge; there was an average decrease of 94% in testosterone at day 3. Most of the leuprorelin-treated patients experienced testosterone surge; there was an average increase of 65% in testosterone at day 3. This difference was statistically significant (p<0.001). (See Figure 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe primary end-point in the study was testosterone suppression rates after one year of treatment with degarelix or leuprorelin. The clinical benefit for degarelix compared to leuprorelin plus anti-androgen in the initial phase of treatment has not been demonstrated.
Testosterone Reversibility: In a study involving patients with rising PSA after localised therapy (mainly radical prostatectomy and radiation) were administered FIRMAGON for seven months followed by a seven months monitoring period. The median time to testosterone recovery (>0.5 ng/mL, above castrate level) after discontinuation of treatment was 112 days (counted from start of monitoring period, i.e 28 days after last injection). The median time to testosterone >1.5 ng/ml (above lower limit of normal range) was 168 days.
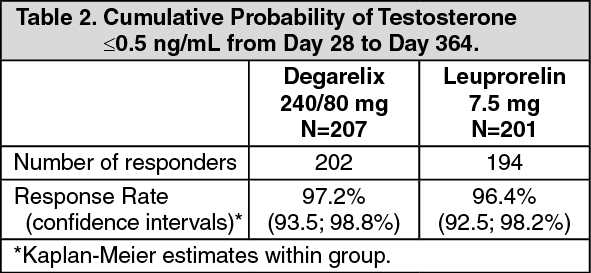
Long-term effect: Successful response in the study was defined as attainment of medical castration at day 28 and maintenance through day 364 where no single testosterone concentration was greater than 0.5 ng/ml. (See Table 2.)
 Click on icon to see table/diagram/image
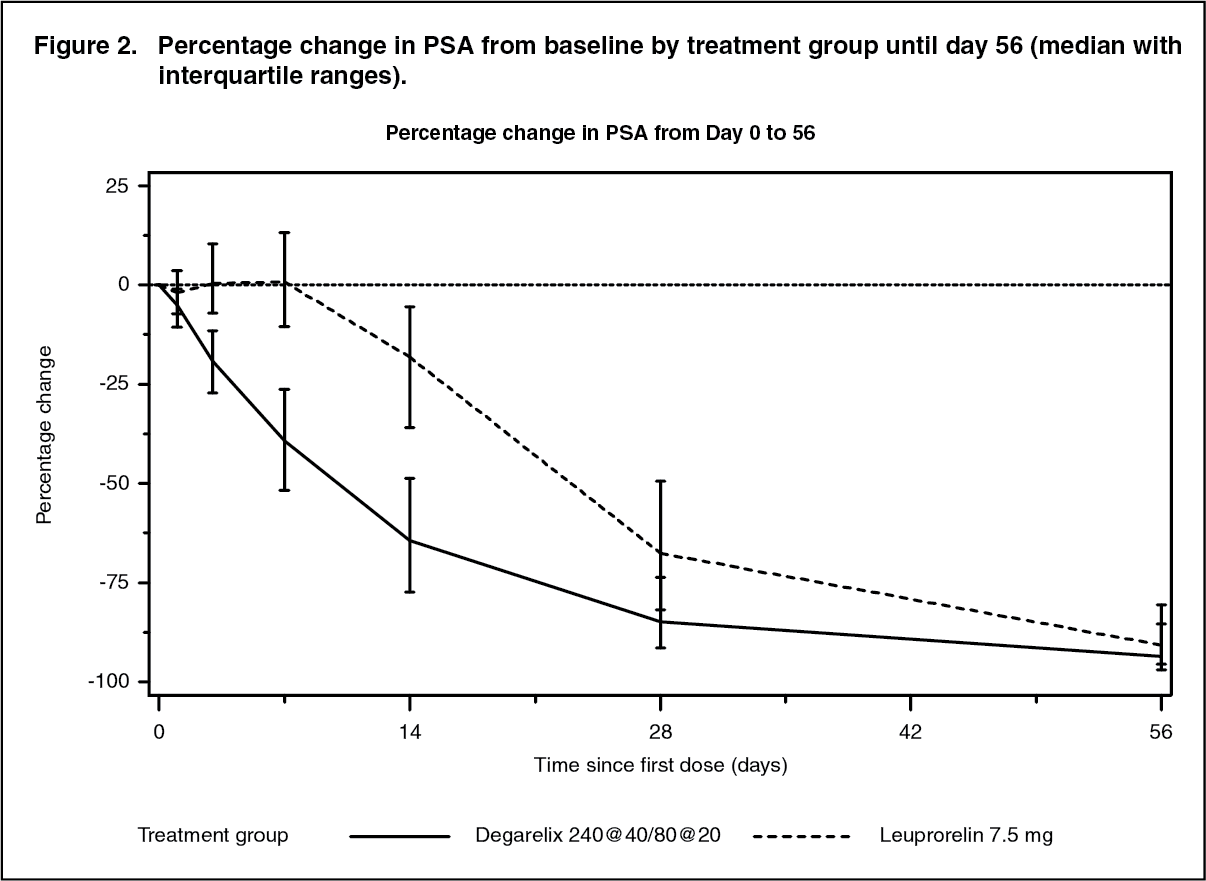
Click on icon to see table/diagram/imageAttainment of prostate specific antigen (PSA) reduction: Tumour size was not measured directly during the clinical trial programme, but there was an indirect beneficial tumour response as shown by a 95% reduction after 12 months in median PSA for degarelix.
The median PSA in the study at baseline was: For the degarelix 240/80 mg treatment group 19.8 ng/ml (interquartile range: P25 9.4 ng/ml, P75 46.4 ng/ml); for the leuprorelin 7.5 mg treatment group 17.4 ng/ml (interquartile range: P25 8.4 ng/ml, P75 56.5 ng/ml). (See Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThis difference was statistically significant (p<0.001) for the pre-specified analysis at day 14 and day 28.
Prostate specific antigen (PSA) levels are lowered by 64% two weeks after administration of degarelix, 85% after one month, 95% after three months, and remained suppressed (approximately 97%) throughout the one year of treatment.
From day 56 to day 364 there were no significant differences between degarelix and the comparator in the percentage change from baseline.
Effect on prostate volume: Three months therapy with degarelix (240/80 mg dose regimen) resulted in a 37% reduction in prostate volume as measured by trans-rectal ultrasound scan (TRUS) in patients requiring hormonal therapy prior to radiotherapy and in patients who were candidates for medical castration. The prostate volume reduction was similar to that attained with goserelin plus anti-androgen protection.
Effect on QT/QTc intervals: In the confirmatory study comparing FIRMAGON to leuprorelin periodic electrocardiograms were performed. Both therapies showed QT/QTc intervals exceeding 450 msec in approximately 20% of the patients. From baseline to end of study the median change for FIRMAGON was 12.0 msec and for leuprorelin it was 16.7 msec.
Anti-degarelix Antibody: Anti-degarelix antibody development has been observed in 10% of patients after treatment with FIRMAGON for one year and 29% of patients after treatment with FIRMAGON for up to 5.5 years. There is no indication that the efficacy or safety of FIRMAGON treatment is affected by antibody formation after up to 5.5 years of treatment.
Pharmacokinetics: Absorption: Following subcutaneous administration of 240 mg degarelix at a concentration of 40 mg/ml to prostate cancer patients in the pivotal study CS21, AUC0-28 days was 635 (602-668) day*ng/mL, Cmax was 66.0 (61.0-71.0) ng/ml and occurred at tmax at 40 (37-42) hours. Mean trough values were approximately 11-12 ng/ml after the starting dose and 11-16 ng/ml after maintenance dosing of 80 mg at a concentration of 20 mg/mL. Cmax degarelix plasma concentration decreases in a biphasic fashion, with a mean terminal half-life (t½) of 29 days for the maintenance dose. The long half-life after subcutaneous administration is a consequence of a very slow release of degarelix from the depot formed at the injection site(s). The pharmacokinetic behavior of the medicinal product is influenced by its concentration in the solution for injection. Thus, Cmax and bioavailability tend to decrease with increasing dose concentration while the half-life is increased. Therefore, no other dose concentrations than the recommended should be used.
Distribution: The distribution volume in healthy elderly men is approximately 1 l/kg. Plasma protein-binding is estimated to be approximately 90%.
Biotransformation: Degarelix is subject to common peptidic degradation during the passage of the hepato-biliary system and is mainly excreted as peptide fragments in the faeces. No significant metabolites were detected in plasma samples after subcutaneous administration. In vitro studies have shown that degarelix is not a substrate for the human CYP450 system.
Elimination: In healthy men, approximately 20-30% of a single intravenously administered dose is excreted in the urine, suggesting that 70-80% is excreted via the hepatobiliary system. The clearance of degarelix when administered as single intravenous doses (0.864-49.4 μg/kg) in healthy elderly men was found to be 35-50 ml/hr/kg.
Special populations: Patients with renal impairment: No pharmacokinetic studies in renally impaired patients have been conducted. Only about 20-30% of a given dose of degarelix is excreted unchanged by the kidneys. A population pharmacokinetics analysis of the data from the confirmatory Phase III study has demonstrated that the clearance of degarelix in patients with mild to moderate renal impairment is reduced by approximately 23%; therefore, dose adjustment in patients with mild or moderate renal impairment is not recommended. Data on patients with severe renal impairment is scarce and caution is therefore warranted in this patient population.
Patients with hepatic impairment: Degarelix has been investigated in a pharmacokinetic study in patients with mild to moderate hepatic impairment. No signs of increased exposure in the hepatically impaired subjects were observed compared to healthy subjects. Dose adjustment is not necessary in patients with mild or moderate hepatic impairment. Patients with severe hepatic dysfunction have not been studied and caution is therefore warranted in this group.
Toxicology: Preclinical safety data: Animal reproduction studies showed that degarelix caused infertility in male animals. This is due to the pharmacological effect; and the effect was reversible.
In female reproduction toxicity studies degarelix revealed findings expected from the pharmacological properties. It caused a dosage dependent prolongation of the time to mating and to pregnancy, a reduced number of corpora lutea and an increase in the number of pre- and post-implantation losses, abortions, early embryo/foetal deaths, premature deliveries and in the duration of parturition.
Nonclinical studies on safety pharmacology, repeated-dose toxicity, genotoxicity and carcinogenic potential revealed no special hazard for humans. Both in vitro and in vivo studies showed no signs of QT prolongation.
No target organ toxicity was observed from acute, subacute and chronic toxicity studies in rats and monkeys following subcutaneous administration of degarelix. Drug-related local irritation was noted in animals when degarelix was administered subcutaneously in high doses.