


 Sign Out
Sign Out


A TTR stabilisation assay was utilised as a pharmacodynamic marker and assessed the stability of the TTR tetramer under denaturation conditions. The TTR stabilisation assay quantifies immunoturbidimetric measurement of the stable TTR tetramer in plasma pre- and post-treatment with 2-day in vitro denaturation with urea. Using this assay, a dose-dependent trend for greater TTR tetramer stabilisation is observed for tafamidis meglumine 80 mg compared to tafamidis meglumine 20 mg. However, the clinical relevance of a higher TTR tetramer stabilisation towards cardiovascular outcomes is not known.
Tafamidis stabilised both the wild-type TTR tetramer and the tetramers of 14 TTR variants tested clinically after once-daily dosing. Tafamidis also stabilised the TTR tetramer for an additional 25 variants tested ex vivo, thus demonstrating TTR stabilisation of 40 amyloidogenic TTR genotypes.
A population PK/PD analysis was conducted with a database consisting of 3662 observations from 102 healthy subjects and 558 patients with transthyretin amyloidosis.
None of the following parameters were found to modify the VYNDAMAX pharmacodynamic response: race (non-Japanese vs. Japanese), patient type (healthy volunteer, ATTR-PN, ATTR-CM), or genotype.
Clinical studies: Efficacy was demonstrated in a multicenter, international, double-blind, placebo-controlled, randomised 3-arm study in 441 patients with wild-type or hereditary ATTR-CM.
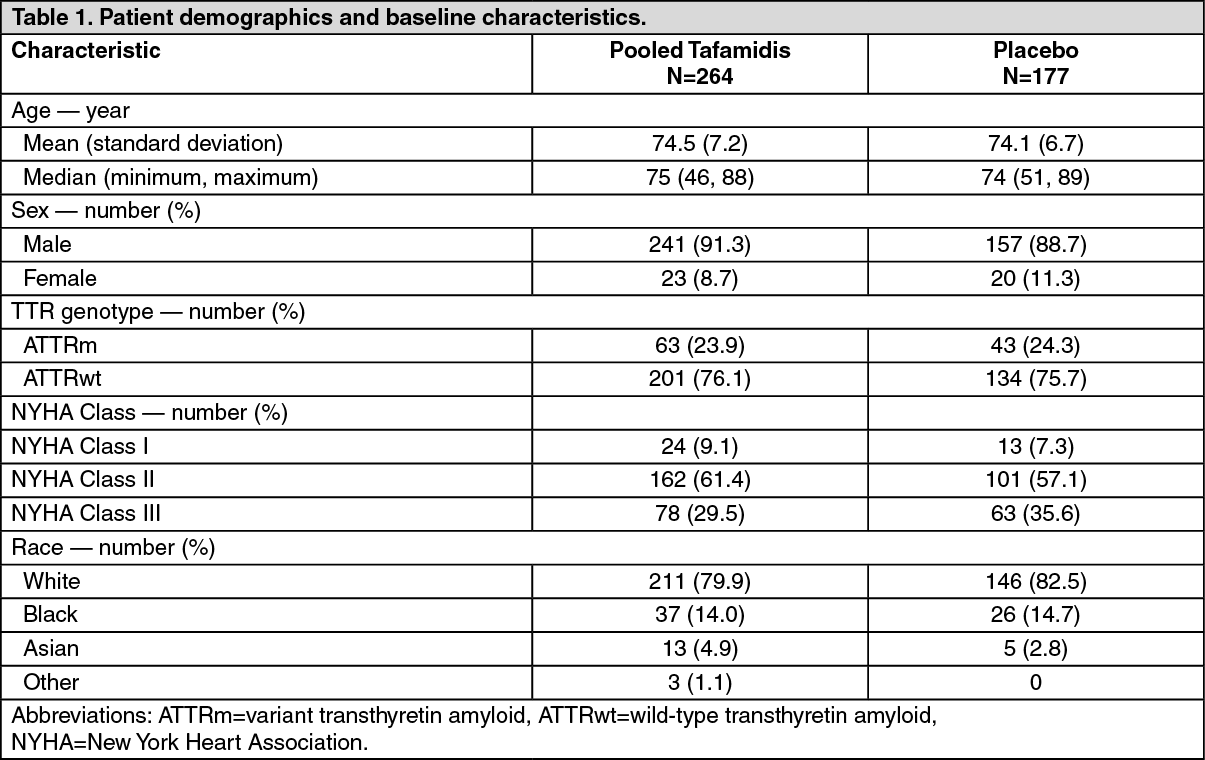
Patients were randomised to either tafamidis meglumine 20 mg (n=88) or 80 mg [administered as four 20 mg tafamidis meglumine capsules] (n=176) or matching placebo (n=177) once daily, in addition to standard of care (e.g., diuretics) for 30 months. Tafamidis meglumine 80 mg is bioequivalent to tafamidis 61 mg (see Pharmacokinetics as follows). Treatment assignment was stratified by the presence or absence of a variant TTR genotype as well as by baseline severity of disease (NYHA Class). Table 1 describes the patient demographics and baseline characteristics. Patients with NYHA Class IV were excluded from the study. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe primary analysis used a hierarchical combination applying the method of Finkelstein-Schoenfeld (F-S) to all-cause mortality and frequency of cardiovascular-related hospitalisations, which is defined as the number of times a subject is hospitalised (i.e., admitted to a hospital) for cardiovascular-related morbidity. The method compared each patient to every other patient within each stratum in a pair-wise manner that proceeded in a hierarchical fashion using all-cause mortality followed by frequency of cardiovascular-related hospitalisations when patients could not be differentiated based on mortality.
This analysis demonstrated a significant reduction (p=0.0006) in all-cause mortality and frequency of cardiovascular-related hospitalisations in the pooled 20 mg and 80 mg tafamidis dose group versus placebo (Table 2). (See Table 2.)
 Click on icon to see table/diagram/image
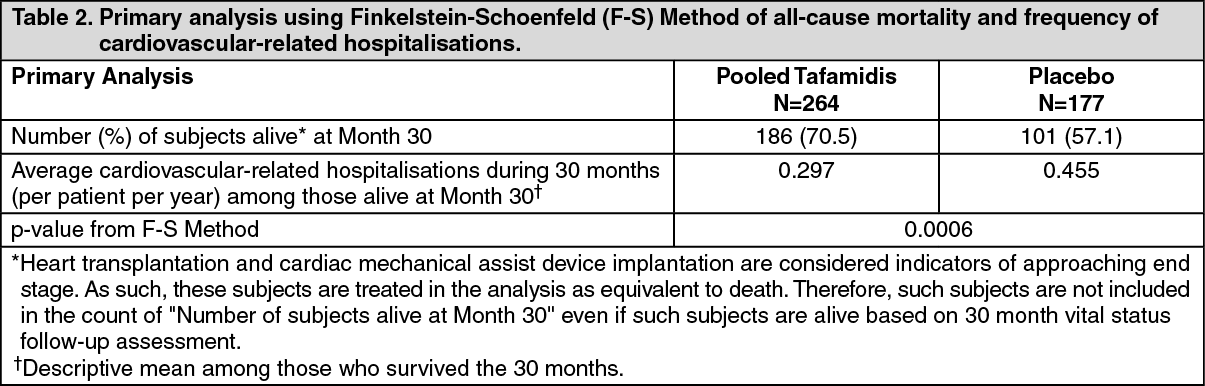
Click on icon to see table/diagram/imageAnalysis of the individual components of the primary analysis (all-cause mortality and cardiovascular-related hospitalisation) also demonstrated significant reductions for tafamidis versus placebo.
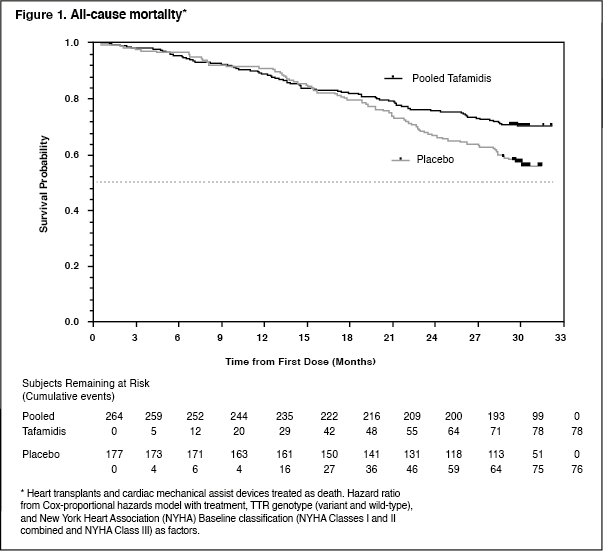
The hazard ratio from the all-cause mortality Cox-proportional hazard model for pooled tafamidis was 0.698 (95% CI 0.508, 0.958), indicating a 30.2% reduction in the risk of death relative to the placebo group (p=0.0259). A Kaplan-Meier plot of time to event all-cause mortality is presented in Figure 1. (See Figure 1.)
 Click on icon to see table/diagram/image
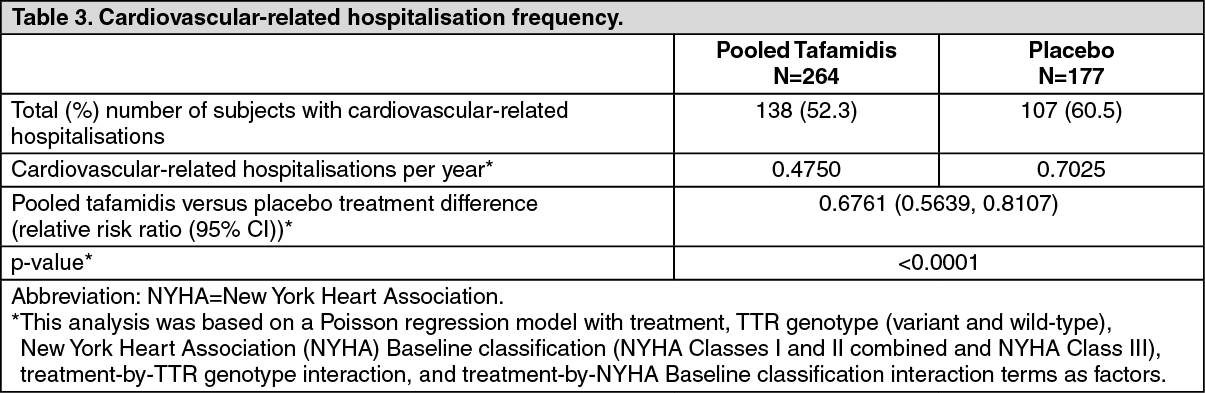
Click on icon to see table/diagram/imageThere were significantly fewer cardiovascular-related hospitalisations with tafamidis compared with placebo with a reduction in risk of 32.4% (Table 3). (See Table 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe treatment effects of tafamidis on functional capacity and health status were assessed by the 6-Minute Walk Test (6MWT) and the Kansas City Cardiomyopathy Questionnaire-Overall Summary (KCCQ-OS) score, respectively. A significant treatment effect favouring tafamidis was first observed at Month 6 and remained consistent through Month 30 on both 6MWT distance and KCCQ-OS score (Figure 2 and Table 4). (See Figure 2.)
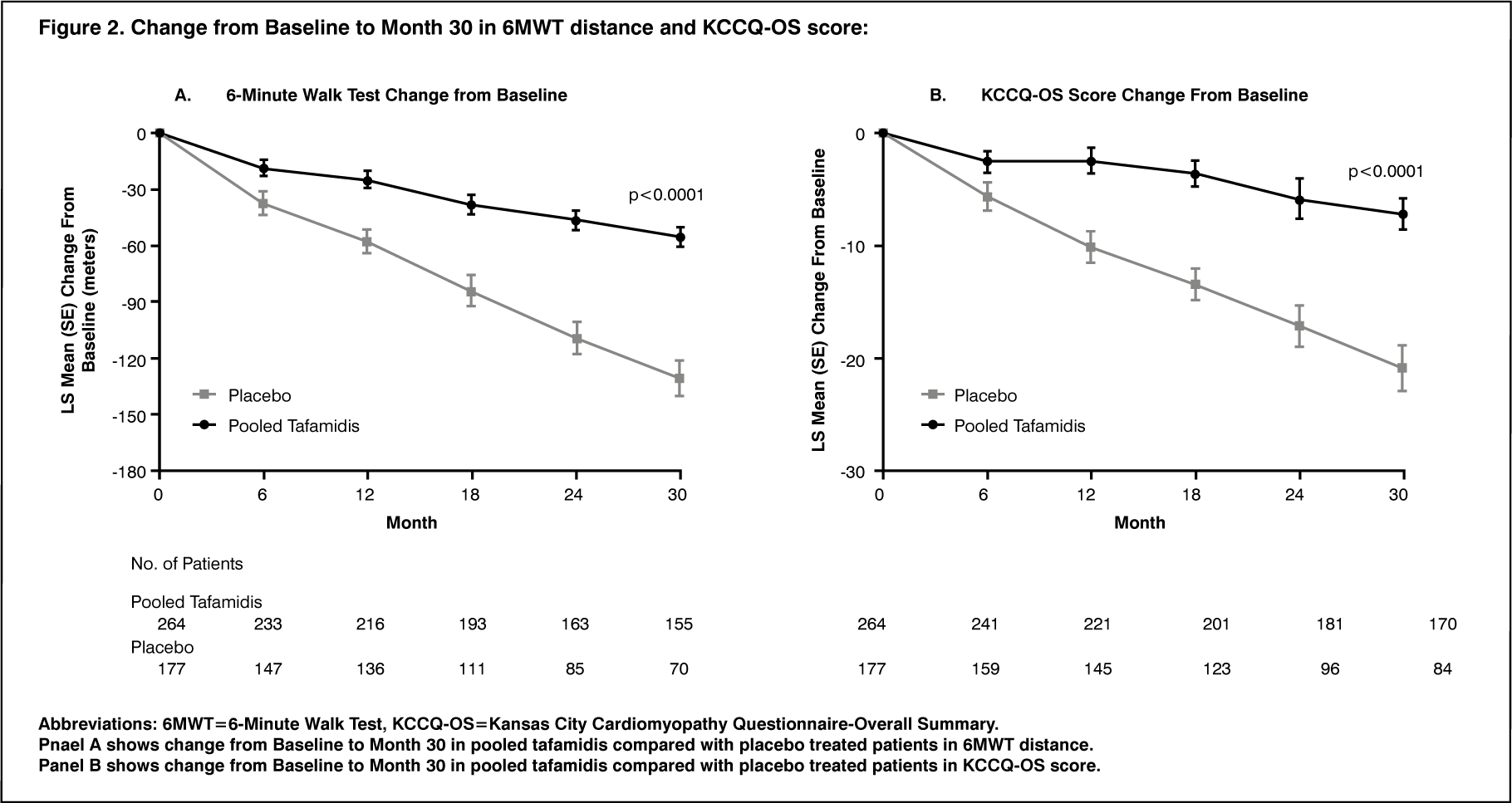 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe KCCQ-OS score is composed of four domains including Total Symptom (Symptom Frequency and Symptom Burden), Physical Limitation, Quality of Life, and Social Limitation. All four domains significantly favoured tafamidis compared to placebo at Month 30 (Figure 2 and Table 4). KCCQ-OS and domain scores range from 0-100 with higher scores representing better health status. The cumulative distribution and distribution for change from Baseline to Month 30 for KCCQ-OS show that the proportion of patients with declining KCCQ-OS scores was lower for the pooled tafamidis treated group compared to placebo (Figure 3). (See Figure 3 and Table 4.)
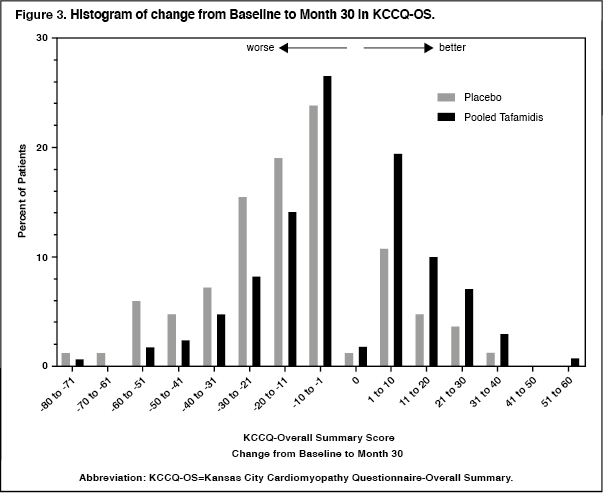 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image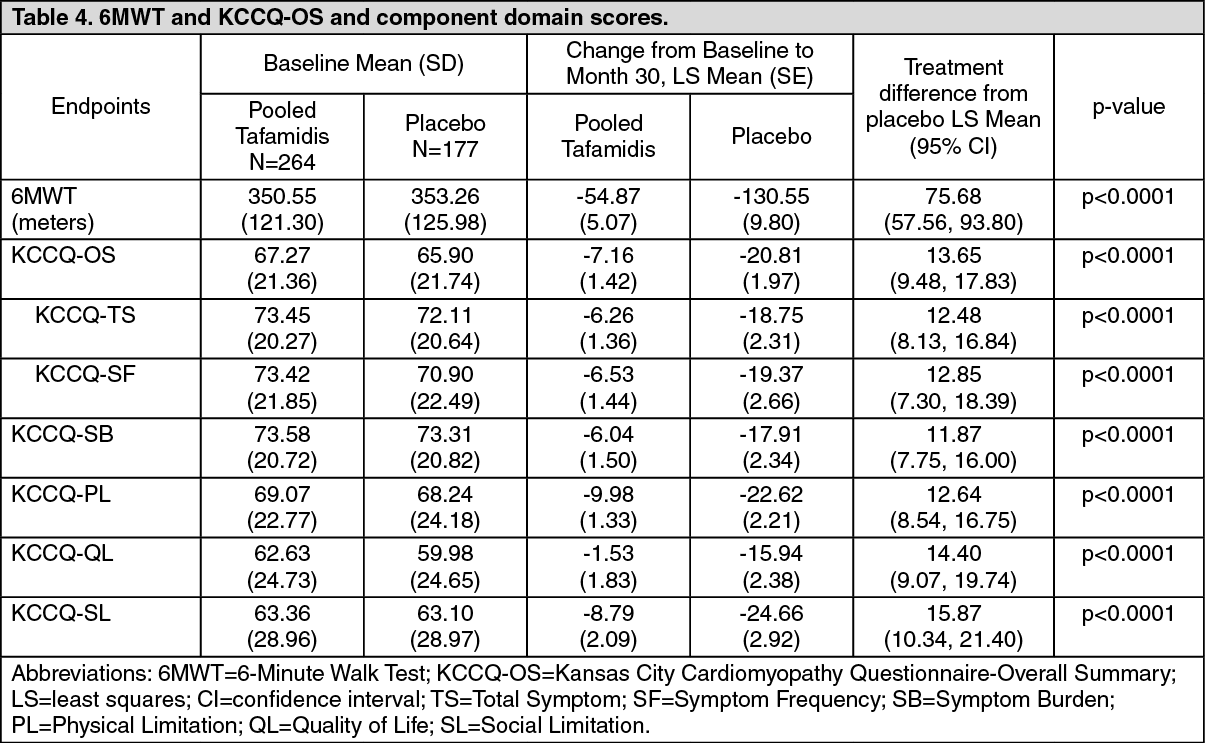 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn the pivotal study, patients who exhibited >32% TTR stabilization were considered stabilised. At Month 1, a significantly greater proportion of patients in the pooled tafamidis group (211/245 [86.1%] patients) demonstrated TTR stabilisation than was observed for patients in the placebo group (6/170 [3.5%] patients) (p<0.0001).
Results from F-S method represented by win ratio for the combined endpoint and its components (all-cause mortality and frequency of cardiovascular-related hospitalisation) consistently favoured tafamidis versus placebo across all subgroups (wild-type, variant and NYHA Class I & II, and III) except for cardiovascular-related hospitalisation frequency in NYHA Class III (Figure 4). Win ratio is the number of pairs of treated-patient "wins" divided by number of pairs of placebo patient "wins". Analyses of 6MWT and KCCQ-OS also favoured tafamidis relative to placebo within each subgroup. (See Figure 4.)
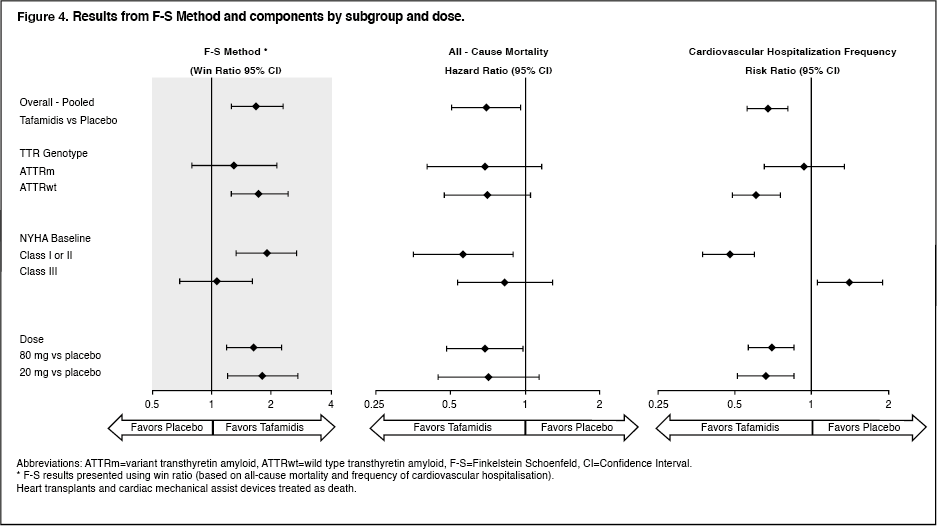 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn applying the F-S method to each dose group individually, tafamidis reduced the combination of all-cause mortality and frequency of cardiovascular-related hospitalisations for both the 80 mg and 20 mg doses compared to placebo (p=0.0030 and p=0.0048, respectively).
Results of the components of the primary analysis, functional capacity and health status (6MWT and KCCQ-OS at Month 30), cardiovascular-related mortality, and TTR stabilisation at Month 1 were analysed by individual doses (80 mg and 20 mg) compared to placebo. The comparison of each dose versus placebo demonstrated an effect with tafamidis for all analyses. The observed results were similar for subjects treated with either the tafamidis meglumine 80 mg or 20 mg doses.
Biomarkers associated with heart failure (NT-proBNP and Troponin I) differentiated between the 80 mg and the 20 mg doses. For NT-proBNP, the LS mean difference in change from Baseline to Month 30 from placebo for 20 mg tafamidis meglumine was -1417.02 pg/mL (SE=743.38) and for 80 mg was -2587.54 pg/mL (SE=570.25). Further, the LS mean difference between the 20 mg and 80 mg doses was 1170.51 pg/mL (SE=587.31) (p=0.0468), favouring the 80 mg dose group. Similar results were observed for Troponin I where the LS mean difference in change from Baseline to Month 30 from placebo for tafamidis meglumine 20 mg was -0.06 ng/mL (SE=0.045) and for 80 mg was -0.10 ng/mL (SE=0.018). The LS mean difference between the 20 mg and 80 mg doses for Troponin I was 0.05 ng/mL (SE=0.04) (p=0.2479), numerically favouring the 80 mg dose group, although the trend did not reach statistical significance.
In an ongoing extension study, patients who completed 30 months of blinded treatment in the pivotal study were eligible to continue to receive 20 mg or 80 mg tafamidis meglumine. Dose blind was maintained during this extension phase. As of February 2018, the median duration of treatment was approximately 36 months. In a post-hoc comparison of all-cause mortality in the extension study by dose, the Hazard Ratio was 0.8976 (95% CI 0.5711, 1.4108), suggesting a 10.2% reduction in risk of death in patients receiving 80 mg relative to patients receiving 20 mg, but this difference was not statistically significant (p=0.6395).
A supra-therapeutic, single, 400 mg oral dose of tafamidis meglumine solution in healthy volunteers demonstrated no effect on the QTc interval.
Pharmacokinetics: The pharmacokinetic profile of VYNDAMAX was determined in Phase I studies in healthy volunteers and patients with ATTR-PN or ATTR-CM.
Absorption: After oral administration of VYNDAMAX once daily, the maximum peak concentration (Cmax) is achieved at a median time (tmax) within 4 hours after dosing in the fasted state. Concomitant administration of a high fat, high calorie meal altered the rate of absorption, but not the extent of absorption. These results support the administration of VYNDAMAX with or without food.
Distribution: Tafamidis is highly protein bound (>99%) in plasma. The apparent steady-state volume of distribution is 16 litres for tafamidis meglumine and 18.5 litres for tafamidis, based on 75 kg adult.
Metabolism and excretion: While there is no explicit evidence of biliary excretion of tafamidis in humans, based on preclinical data, it is suggested that tafamidis is metabolised by glucuronidation and excreted via the bile. This route of metabolism and excretion is likely in humans, as approximately 59% of the total administered dose is recovered in faeces mostly as unchanged drug, and approximately 22% recovered in urine mostly as the glucuronide metabolite. Based on population pharmacokinetic results, the apparent oral clearance of tafamidis meglumine is 0.228 L/h (0.263 L/h for tafamidis) and the population mean half-life is approximately 49 hours.
Dose and time linearity: Exposure from once-daily dosing with tafamidis meglumine increased with increasing dose up to 480 mg single dose and multiple doses up to 80 mg per day. In general, increases were proportional or near proportional to dose.
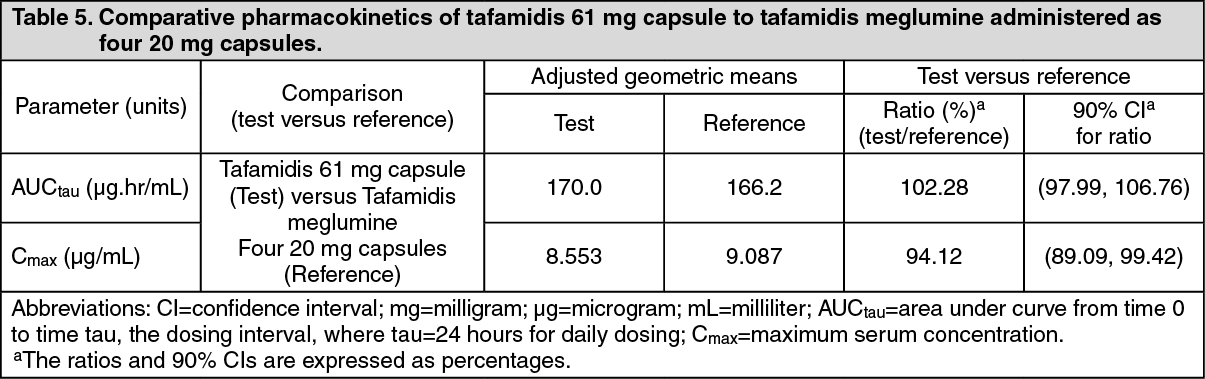
Tafamidis 61 mg provides steady-state exposures (Cmax and AUC) equivalent to 80 mg tafamidis meglumine (administered as four 20 mg capsules), which was administered to patients with ATTR-CM in the double-blind, placebo-controlled, randomised study (Table 5) (see Pharmacodynamics as previously mentioned). (See Table 5.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageMean half-life and oral clearance were similar after single and repeated administration of a 20 mg dose of tafamidis meglumine, indicating a lack of induction or inhibition of tafamidis metabolism.
Results of once-daily dosing with tafamidis meglumine 15 mg to 60 mg oral solution for 14 days demonstrated that steady-state (ss) was achieved by Day 14.
Drug interactions: No significant effect was observed on the pharmacokinetics of midazolam (a CYP3A4 substrate) or on the formation of its active metabolite (1-hydroxymidazolam), when a single 7.5 mg dose of midazolam was administered prior to and after a 14-day regimen of 20 mg once-daily tafamidis meglumine. The overall systemic exposure (AUC0-∞) and total clearance (CL/F) of midazolam were shown to be equivalent. In addition, tafamidis did not induce CYP3A4 activity in either male or female subjects.
Pharmacokinetics in special patient groups: Elderly patients: Based on population pharmacokinetic results, patients ≥65 years of age had an average 15% lower estimate of apparent oral clearance at steady-state when compared with patients under the age of 65. However, the difference in clearance results in <20% increases in mean Cmax and AUC compared to younger subjects and is not clinically significant.
Renally impaired patients: VYNDAMAX has not specifically been evaluated in patients with renal impairment. Tafamidis is primarily metabolised by glucuronidation and is likely excreted via the hepatobiliary pathway. The influence of creatinine clearance on tafamidis pharmacokinetics (PK) was evaluated in a population PK analysis in patients with creatinine clearance >18 mL/min. Pharmacokinetic estimates indicated no difference in apparent oral clearance of tafamidis in patients with creatinine clearance <80 mL/min compared to those with creatinine clearance ≥80 mL/min. No dosage adjustment is required in patients with renal impairment. Limited data are available in patients with severe renal impairment (creatinine clearance ≤30 mL/min).
Hepatically impaired patients: No dose adjustment is necessary in mild or moderate hepatic impairment. Pharmacokinetic data indicated decreased systemic exposure (approximately 40%) and increased total clearance (0.52 L/h versus 0.31 L/h) of tafamidis meglumine in subjects with moderate hepatic impairment (Child-Pugh Score of 7-9 inclusive) compared to healthy subjects. As TTR levels are lower in patients with moderate hepatic impairment than in healthy subjects, the exposure of VYNDAMAX relative to the amount of TTR would be sufficient for stabilisation of the TTR tetramer in these patients. Exposure to VYNDAMAX was similar between subjects with mild hepatic impairment and healthy subjects.
The exposure to VYNDAMAX in patients with severe hepatic impairment is unknown.
Toxicology: Nonclinical safety data: Fertility: There were no effects of tafamidis meglumine on fertility, reproductive performance, or mating behaviour in the rat at any dose. Rats were dosed daily (5, 15, and 30 mg/kg/day) prior to cohabitation (for at least 15 days for females and 28 days for males), throughout the cohabitation period to the day prior to termination of males and through to implantation of females (Gestation Day 7). No adverse effects were noted on male rats in toxicity, fertility and mating behaviour at any dose. Because no reproductive effects occurred at the highest dose tested, the paternal and maternal no observed effect level for reproductive toxicity of tafamidis meglumine is greater than 30 mg/kg/day (9.5 times the clinical AUC at the maximum recommended human dose [MRHD] of 61 mg tafamidis per day, and estimated to be greater than 9.7 times the clinical AUC at the MRHD of 80 mg tafamidis meglumine per day).
Developmental toxicity: In pregnant rabbits increased skeletal variations were observed at ≥0.5 mg/kg/day (exposures approximately equivalent to the clinical exposures at the MRHD of 61 mg tafamidis and 80 mg tafamidis meglumine respectively), while increased skeletal malformations, reduced embryo-fetal survival and reduction in fetal body weights were observed at 8 mg/kg/day (AUC exposures ≥9.1 times and 9.3 times clinical AUC at the MRHD of 61 mg tafamidis and 80 mg tafamidis meglumine respectively). In pregnant rats, oral administration of tafamidis (15, 30, and 45 mg/kg/day) from Gestation Day 7 through 17 resulted in decreased fetal weights at ≥30 mg/kg/day (approximately ≥9.5 times and ≥9.7 times the human AUC at the clinical dose of 61 mg tafamidis and 80 mg tafamidis meglumine respectively).
In the rat pre- and post-natal development study with tafamidis, pregnant rats were orally administered tafamidis meglumine at doses of 5, 15, or 30 mg/kg/day from Gestation Day 7 through Lactation Day 20. Decreased pup survival, reduced pup weights and malformations (microphthalmia, enophthalmos, domed head) were noted at doses ≥15 mg/kg/day (≥6.4 times and ≥6.6 times the clinical AUC at the MRHD of 61 mg tafamidis and 80 mg tafamidis meglumine per day respectively). Decreased pup weights in males were associated with delayed sexual maturation (preputial separation) at 15 mg/kg/day. Impaired performance in a water-maze test for learning and memory was observed at 15 mg/kg/day. The no observable adverse effect level (NOAEL) for viability and growth in the F1 generation offspring following maternal exposures to tafamidis was 5 mg/kg/day (human equivalent dose of 0.8 mg/kg/day), a dose approximately 0.92 times and 1.2 times the clinical dose of 61 mg tafamidis and 80 mg tafamidis meglumine respectively for a 70 kg adult.
Genotoxicity: Tafamidis was not genotoxic in a bacterial reverse mutation assay, in an in vitro human lymphocyte chromosomal aberration assay or in an in vivo rat bone marrow micronucleus test. Nonclinical data demonstrated no special hazard for humans based on conventional studies of genotoxicity.
Carcinogenicity: There was no evidence of increased incidence of neoplasia in a 2-year carcinogenicity study in rats at exposures up to 18 times the human AUC at the clinical doses of 61 mg tafamidis and 80 mg tafamidis meglumine. There was no evidence of an increased incidence of neoplasia in the transgenic (Tg)-rasH2 mouse following repeat daily administration for 26 weeks at exposures up to 9.6 times and 9.9 times the human AUC at the clinical doses of 61 mg tafamidis and 80 mg tafamidis meglumine respectively. In this study, significant non neoplastic lesions were noted in the kidneys (nephrosis) and liver (centrilobular hypertrophy and single cell necrosis) in the Tg-rasH2 mice at dose levels ≥2.8 times and ≥2.9 times the clinical AUC at 61 mg tafamidis and 80 mg tafamidis meglumine respectively.