Pharmacotherapeutic Group: Not assigned, combinations.
ATC Code: Not assigned.
Pharmacology: Pharmacodynamics: Lamivudine/Tenofovir Disoproxil Fumarate: Mechanism of action and Pharmacodynamic Effects: Lamivudine, the negative enantiomer of 2'-deoxy-3'-thiacytidine, is a dideoxynucleoside analogue. Tenofovir disoproxil fumarate is converted
in vivo to tenofovir, a nucleoside monophosphate (nucleotide) analogue of adenosine monophosphate.
Lamivudine and tenofovir are phosphorylated by cellular enzymes to form lamivudine triphosphate and tenofovir diphosphate, respectively. Lamivudine triphosphate and tenofovir diphosphate competitively inhibit HIV-1 reverse transcriptase (RT), resulting in DNA chain termination. Both substances are active against HIV-1 and HIV-2, as well as against hepatitis B virus.
Resistance: In many cases when a lamivudine-containing treatment regimen fails, the M184V mutation will be selected for at an early stage. M184V causes high-level resistance to lamivudine (>300-fold reduced susceptibility). Virus with M184V replicates less well than does wild type virus. M184V causes high-level resistance to lamivudine (>300-fold reduced susceptibility).
In vitro data tend to suggest that the continuation of lamivudine in an antiretroviral regimen despite the development of M184V might provide residual anti-retroviral activity (likely through impaired viral fitness). The clinical relevance of these findings is not established.
Cross-resistance conferred by the M184V mutation is limited within the nucleoside/nucleotide inhibitor class of antiretroviral agents. M184V confers full cross-resistance against emtricitabine. Zidovudine and stavudine maintain their antiretroviral activities against lamivudine-resistant HIV-1. Abacavir maintains its antiretroviral activities against lamivudine-resistant HIV-1 harbouring only the M184V mutation. The M184V mutant shows a <4-fold decrease in susceptibility to didanosine; the clinical significance of this is unknown.
The K65R mutation is selected
in vitro when HIV-1 is cultured in the presence of increasing tenofovir concentrations. It may also emerge
in vivo upon virological failure of a treatment regimen including tenofovir. K65R reduces tenofovir susceptibility
in vitro approximately 2-fold, and has been associated with a lack of response to tenofovir-containing regimens.
Clinical studies in treatment-experienced patients have assessed the anti-HIV activity of tenofovir against strains of HIV-1 with thymidine analogue mutations (TAMs), which are not selected for by tenofovir. Patients whose HIV expressed 3 or more TAMs that included either the M41L or L210W mutation showed reduced response to tenofovir.
Clinical Efficacy: When tenofovir disoproxil fumarate and lamivudine were combined with efavirenz in treatment-naïve patients with HIV-1, the proportion of patients (ITT) with HIV-RNA <50 copies/ml were 76.3% and 67.8% at 48 and 144 weeks, respectively.
No specific studies with the combination tenofovir disoproxil fumarate, Lamivudine and dolutegravir have been conducted in adolescents.
Dolutegravir: Mechanism of action: Dolutegravir inhibits HIV integrase by binding to the integrase active site and blocking the strand transfer step of retroviral Deoxyribonucleic acid (DNA) integration which is essential for the HIV replication cycle.
Pharmacodynamic Effects: Antiviral activity in cell culture: The IC
50 for dolutegravir in various labstrains using PBMC was 0.5 nM, and when using MT-4 cells it ranged from 0.7-2 nM. Similar IC
50s were seen for clinical isolates without any major difference between subtypes; in a panel of 24 HIV-1 isolates of clades A, B, C, D, E, F and G and group O the mean IC
50 value was 0.2 nM (range 0.02-2.14). The mean IC
50 for 3 HIV-2 isolates was 0.18 nM (range 0.09-0.61).
Antiviral activity in combination with other antiviral agents: No antagonistic effects in vitro were seen with dolutegravir and other antiretrovirals tested: stavudine, abacavir, efavirenz, nevirapine, lopinavir, amprenavir, enfuvirtide, maraviroc and raltegravir. In addition, no antagonistic effects were seen for dolutegravir and adefovir, and ribavirin had no apparent effect on dolutegravir activity.
Effect of human serum: In 100% human serum, the mean protein fold shift was 75 fold, resulting in protein adjusted IC90 of 0.064 ug/mL.
Resistance: Resistance
in vitro:
Serial passage is used to study resistance evolution
in vitro. When using the lab-strain HIV-1 IIIB during passage over 112 days, mutations selected appeared slowly, with substitutions at positions S153Y and F, resulting in a maximal fold change in susceptibility of 4 (range 2-4). These mutations were not selected in patients treated with dolutegravir in the clinical studies. Using strain NL432, mutations E92Q (FC 3) and G193E (also FC 3) were selected. The E92Q mutation has been selected in patients with pre-existing raltegravir resistance who were then treated with dolutegravir (listed as a secondary mutation for dolutegravir).
In further selection experiments using clinical isolates of subtype B, mutation R263K was seen in all five isolates (after 20 weeks and onwards). In subtype C (n=2) and A/G (n=2) isolates the integrase substitution R263K was selected in one isolate, and G118R in two isolates. R263K was reported from two ART experienced, INI naive individual patients with subtypes B and C in the clinical program, but without effects on dolutegravir susceptibility
in vitro. G118R lowers the susceptibility to dolutegravir in site directed mutants (FC 10), but was not detected in patients receiving dolutegravir in the Phase III program.
Primary mutations for raltegravir/elvitegravir (Q148H/R/K, N155H, Y143R/H/C, E92Q and T66I) do not affect the
in vitro susceptibility of dolutegravir as single mutations. When mutations listed as secondary integrase inhibitor associated mutations (for raltegravir/elvitegravir) are added to these primary mutations in experiments with site directed mutants, dolutegravir susceptibility is still unchanged (FC <2 vs wild type virus), except in the case of Q148-mutations, where a FC of 5-10 or higher is seen with combinations of certain secondary mutations. The effect by the Q148-mutations (H/R/K) was also verified in passage experiments with site directed mutants. In serial passage with strain NL432, starting with site directed mutants harbouring N155H or E92Q, no further selection of resistance was seen (FC unchanged around 1). In contrast, starting with mutants harbouring mutation Q148H (FC 1), a variety of secondary mutations were seen with a consequent increase of FC to values >10.
A clinically relevant phenotypic cut-off value (FC vs wild type virus) has not been determined; genotypic resistance was a better predictor for outcome.
Seven hundred and five raltegravir resistant isolates from raltegravir experienced patients were analyzed for susceptibility to dolutegravir. Dolutegravir has a less than or equal to 10 FC against 94% of the 705 clinical isolates.
Resistance
in vivo: In previously untreated patients receiving dolutegravir + 2 NRTIs in Phase IIb and Phase III, no development of resistance to the integrase class, or to the NRTI class was seen (n=1118 follow-up of 48-96 weeks).
In patients with prior failed therapies, but naïve to the integrase class (SAILING study), integrase inhibitor substitutions were observed in 4/354 patients (follow-up 48 weeks treated with dolutegravir, which was given in combination with an investigator selected background regimen (BR). Of these four, two subjects had a unique R263K integrase substitution, with a maximum FC of 1.93, one subject had a polymorphic V151V/I integrase substitution, with maximum FC of 0.92, and one subject had pre-existing integrase mutations and is assumed to have been integrase experienced or infected with integrase resistant virus by transmission. The R263K mutation was also selected
in vitro (see above).
In the presence of integrase class-resistance (VIKING-3 study) the following mutations were selected in 32 patients with protocol defined virological failure (PDVF) through Week 24 and with paired genotypes (all treated with dolutegravir 50 mg twice daily + optimized background agents): L74L/M (n=1), E92Q (n=2), T97A (n=9), E138K/A/T (n=8), G140S (n=2), Y143H (n=1), S147G (n=1), Q148H/K/R (n=4), and N155H (n=1) and E157E/Q (n=1). Treatment emergent integrase resistance typically appeared in patients with a history of the Q148-mutation (baseline or historic). Five further subjects experienced PDVF between weeks 24 and 48, and 2 of these 5 had treatment emergent mutations. Treatment-emergent mutations or mixtures of mutations observed were L74I (n=1), N155H (n=2).
The VIKING-4 study examined dolutegravir (plus optimized background therapy) in subjects with primary genotypic resistance to INIs at Screening in 30 subjects. Treatment-emergent mutations observed were consistent with those observed in the VIKING-3 study.
Effects on electrocardiogram: No relevant effects were seen on the QTc interval, with doses exceeding the clinical dose by approximately three fold.
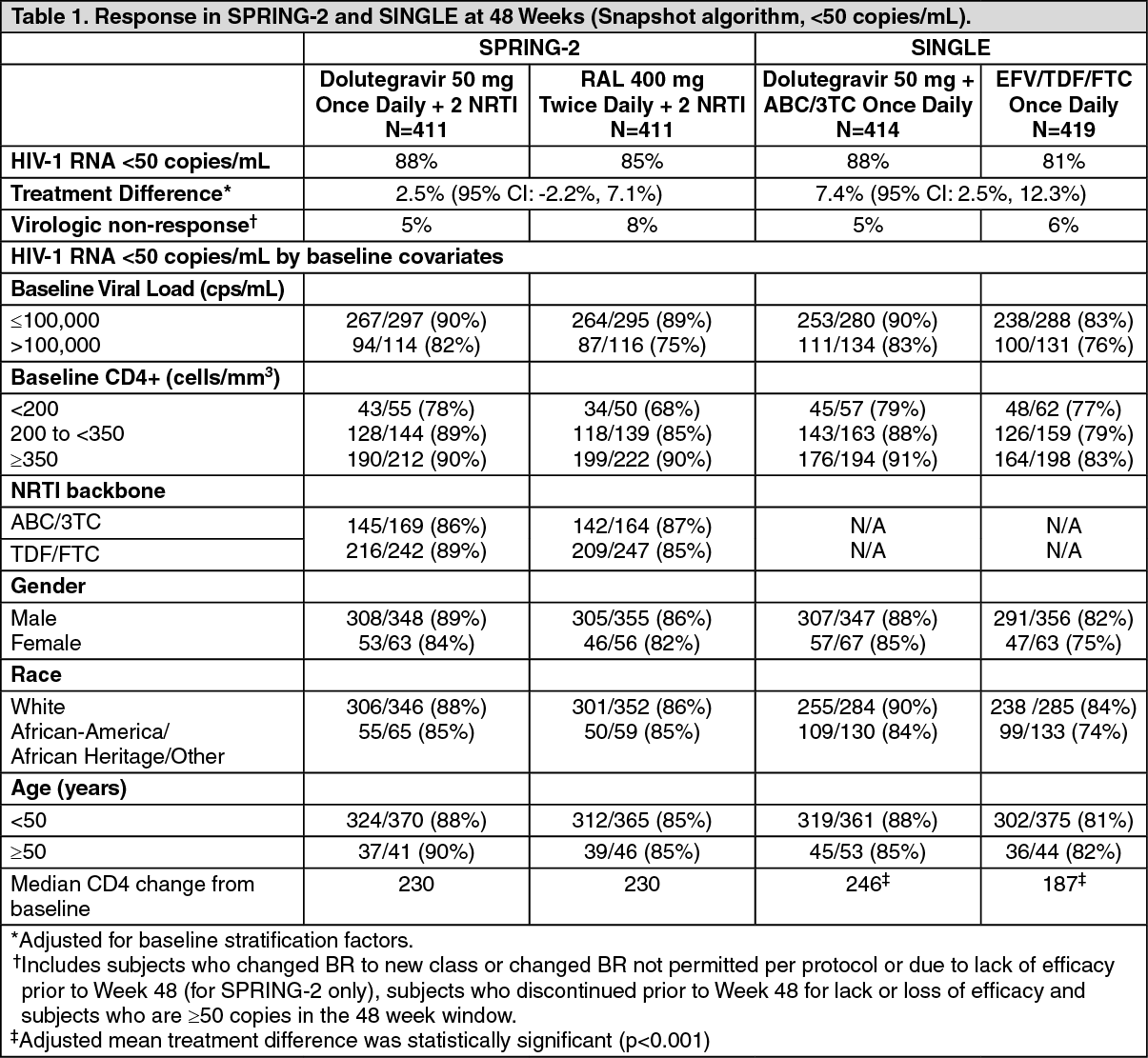
Clinical Efficacy and Safety: Previously untreated patients: The efficacy of dolutegravir in HIV-infected, therapy naïve subjects is based on the analyses of 96-week data from two randomized, international, double-blind, active-controlled trials, SPRING-2 (ING113086) and SINGLE (ING114467). This is supported by 96 week data from an open-label, randomized and activecontrolled study FLAMINGO (ING114915) and additional data from the open-label phase of SINGLE to 144 weeks.
In SPRING-2, 822 adults were randomized and received at least one dose of either dolutegravir 50 mg once daily or raltegravir (RAL) 400 mg twice daily, both administered with either ABC/3TC or TDF/FTC. At baseline, median patient age was 36 years, 14% were female, 15% non-white, 11% had hepatitis B and/or C co-infection and 2% were CDC Class C, these characteristics were similar between treatment groups.
In SINGLE, 833 subjects were randomized and received at least one dose of either dolutegravir 50 mg once daily with fixed-dose abacavir-lamivudine (DTG + ABC/3TC) or fixed-dose efavirenz-tenofovir emtricitabine (EFV/TDF/FTC). At baseline, median patient age was 35 years, 16% were female, 32% nonwhite, 7% had hepatitis C co-infection and 4% were CDC Class C, these characteristics were similar between treatment groups.
The primary endpoint and other week 48 outcomes (including outcomes by key baseline covariates) for SPRING-2 and SINGLE are shown in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
At week 48, dolutegravir was non-inferior to raltegravir in the SPRING-2 study, and in the SINGLE study dolutegravir + ABC/3TC was superior to efavirenz/TDF/FTC (p=0.003), Table 1 above. In SINGLE, the median time to viral suppression was shorter in the dolutegravir treated patients (28 vs 84 days, (p<0.0001, analysis pre-specified and adjusted for multiplicity).
At week 96, results were consistent with those seen at week 48. In SPRING-2, dolutegravir was still noninferior to raltegravir (viral suppression in 81% vs 76% of patients), and with a median change in CD4 count of 276 vs 264 cells/mm
3, respectively. In SINGLE, dolutegravir + ABC/3TC was still superior to EFV/TDF/FTC (viral suppression in 80% vs 72%, treatment difference 8.0% (2.3, 13.8), p=0.006, and with an adjusted mean change in CD4 count of 325 vs 281 cells/ mm
3, respectively. At 144 weeks in the open label phase of SINGLE, virologic suppression was maintained, the dolutegravir + ABC/3TC arm (71%) was superior to the EFV/TDF/FTC arm (63%), treatment difference was 8.3% (2.0, 14.6).
In FLAMINGO (ING114915), an open-label, randomised and active-controlled study, 484 HIV-1 infected antiretroviral naïve adults received one dose of either dolutegravir 50 mg once daily (n=242) or darunavir/ritonavir (DRV/r) 800 mg/100 mg once daily (n=242), both administered with either ABC/3TC or TDF/FTC. At baseline, median patient age was 34 years, 15% were female, 28% non-white, 10% had hepatitis B and/or C co-infection, and 3% were CDC Class C; these characteristics were similar between treatment groups. Virologic suppression (HIV-1 RNA <50 copies/mL) in the dolutegravir group (90%) was superior to the DRV/r group (83%) at 48 weeks. The adjusted difference in proportion and 95% CI were 7.1% (0.9, 13.2), p=0.025. At 96 weeks, virologic suppression in the dolutegravir group (80%) was superior to the DRV/r group (68%), (adjusted treatment difference [DTG-(DRV+RTV)]: 12.4%; 95% CI: [4.7, 20.2].
Treatment emergent resistance in previously untreated patients failing therapy: Through 96 weeks in SPRING-2 and FLAMINGO and 144 weeks in SINGLE, no cases of treatment emergent primary resistance to the integrase- or NRTI-class were seen in the dolutegravir-containing arms. For the comparator arms, the same lack of treatment emergent resistance was also the case for patients treated with darunavir/r in FLAMINGO. In SPRING-2, four patients in the RAL-arm failed with major NRTI mutations and one with raltegravir resistance; in SINGLE, six patients in the EFV/TDF/FTC-arm failed with mutations associated with NNRTI resistance, and one developed a major NRTI mutation.
Patients with prior treatment failure, but not exposed to the integrase class: In the international multicentre, double-blind SAILING study (ING111762), 719 HIV-1 infected, antiretroviral therapy (ART)-experienced adults were randomized and received either dolutegravir 50 mg once daily or raltegravir 400 mg twice daily with investigator selected background regimen consisting of up to 2 agents (including at least one fully active agent). At baseline, median patient age was 43 years, 32% were female, 50% non-white, 16% had hepatitis B and/or C co-infection, and 46% were CDC Class C. All patients had at least two class ART resistance, and 49% of subjects had at least 3-class ART resistance at baseline.
Week 48 outcomes (including outcomes by key baseline covariates) for SAILING are shown in Table 2.
Click on icon to see table/diagram/image
In the SAILING study, virologic suppression (HIV-1 RNA <50 copies/mL) in the Dolutegravir arm (71%) was statistically superior to the raltegravir arm (64%), at Week 48 (p=0.03).
Statistically fewer subjects failed therapy with treatment-emergent integrase resistance on Dolutegravir (4/354, 1%) than on raltegravir (17/361, 5%) (p=0.003) (refer to section 'Resistance in vivo' above for details).
Paediatric population: In a Phase I/II 48 week multicentre, open-label study (P1093/ING112578), the pharmacokinetic parameters, safety, tolerability and efficacy of Dolutegravir will be evaluated in combination regimens in HIV-1 infected adolescents.
At 24 weeks, 16 of 23 (70%) adolescents (12 to less than 18 years of age) treated with Dolutegravir once daily (35 mg n=4, 50 mg n=19) plus OBR achieved viral load <50 copies/mL. Four subjects had virologic failure none of which had INI resistance at the time of virologic failure.
Pharmacokinetics: Lamivudine: Absorption and Bioavailability: Lamivudine is rapidly absorbed following oral administration. Bioavailability is between 80 and 85%. Following single dose administration of one tablet of Efavirenz/Lamivudine/Tenofovir Disoproxil Fumarate 600 mg/300 mg/300 mg Tablets in healthy volunteers, the mean (±SD) lamivudine C
max value was 2483 (±706) ng/ml and the corresponding value for AUC was 13457 (±3717) ng.h/ml. The mean (±SD) lamivudine t
max value was 1.92 (±0.93) hours.
Co-administration of lamivudine with food results in a delay of t
max and a lower C
max (decreased by 47%). However, the extent (based on the AUC) of lamivudine absorbed is not influenced.
Distribution: Intravenous studies with lamivudine showed that the mean apparent volume of distribution is 1.3 l/kg. Lamivudine exhibits linear pharmacokinetics over the therapeutic dose range and displays limited binding to the major plasma protein albumin (< 36% serum albumin
in vitro).
Metabolism: Metabolism of lamivudine is a minor route of elimination. Lamivudine is predominantly cleared unchanged by renal excretion. The likelihood of metabolic drug interactions with lamivudine is low due to the small extent of hepatic metabolism (5-10%) and low plasma protein binding.
Elimination: The observed lamivudine half-life of elimination is 5 to 7 hours. The half-life of intracellular lamivudine triphosphate has been estimated to approximately 22 hours. The mean systemic clearance of lamivudine is approximately 0.32 l/h/kg, with predominantly renal clearance (> 70%), including tubular secretion through the organic cationic transport system.
Special Populations: Renal impairment: Studies in patients with renal impairment show that lamivudine elimination is affected by renal dysfunction. Dose reduction is recommended for patients with creatinine clearance ≤50 ml/min (see Dosage & Administration).
Tenofovir disoproxil fumarate: Tenofovir disoproxil fumarate is a water-soluble ester prodrug, which is rapidly converted in vivo to tenofovir and formaldehyde. Tenofovir is converted intracellularly to tenofovir monophosphate and to the active component, tenofovir diphosphate.
Absorption: Following oral administration of tenofovir disoproxil fumarate to HIV infected patients, tenofovir disoproxil fumarate is rapidly absorbed and converted to tenofovir. The oral bioavailability of tenofovir from tenofovir disoproxil fumarate in fasted patients was approximately 25%. Administration of tenofovir disoproxil fumarate with a high fat meal enhanced the oral bioavailability, with an increase in tenofovir AUC by approximately 40% and C
max by approximately 14%.
Following single dose administration of one tablet of Efavirenz/Lamivudine/Tenofovir Disoproxil Fumarate 600 mg/300 mg/300 mg Tablets in healthy volunteers, the mean (±SD) tenofovir C
max value was 277 (±79) ng/ml and the corresponding value for AUC was 2358 (±627) ng.h/ml. The mean (±SD) tenofovir tmax value was 1.17 (±0.57) hours.
Distribution : Following intravenous administration, the steady-state volume of distribution of tenofovir was estimated to be approximately 800 ml/kg.
In vitro protein binding of tenofovir to plasma or serum protein was less than 0.7 and 7.2%, respectively, over the tenofovir concentration range 0.01 to 25 μg/ml.
Elimination: Tenofovir is primarily excreted by the kidney, both by filtration and an active tubular transport system with approximately 70-80% of the dose excreted unchanged in urine following intravenous administration. Total clearance has been estimated to be approximately 230 ml/h/kg (approximately 300 ml/min). Renal clearance has been estimated to be approximately 160 ml/h/kg (approximately 210 ml/min), which is in excess of the glomerular filtration rate. This indicates that active tubular secretion is an important part of the elimination of tenofovir. Following oral administration the terminal half-life of tenofovir is approximately 12 to 18 hours.
Studies have established the pathway of active tubular secretion of tenofovir to be influx into proximal tubule cell by the human organic anion transporters (hOAT) 1 and 3 and efflux into the urine by the multidrug resistant protein 4 (MRP 4).
In vitro studies have determined that neither tenofovir disoproxil fumarate nor tenofovir are substrates for the CYP450 enzymes.
Age and gender: Limited data on the pharmacokinetics of tenofovir in women indicate no major gender effect. Tenofovir exposure achieved in adolescent patients receiving oral daily doses of tenofovir 300 mg was similar to exposures achieved in adults receiving once-daily doses of tenofovir 300 mg. Pharmacokinetic studies have not been performed in children or in the elderly (over 65 years). Pharmacokinetics have not been specifically studied in different ethnic groups.
Renal impairment: Pharmacokinetic parameters of tenofovir were determined following administration of a single dose of tenofovir disoproxil fumarate 300 mg to 40 non-HIV, non-HBV infected patients with varying degrees of renal impairment defined according to baseline creatinine clearance (CrCl) (normal renal function when CrCl > 80 ml/min; mild with CrCl = 50-79 ml/min; moderate with CrCl = 30-49 ml/min and severe with CrCl = 10-29 ml/min). Compared with patients with normal renal function, the mean (%CV) tenofovir exposure increased from 2,185 (12%) ng·h/ml in subjects with CrCl > 80 ml/min to respectively 3,064 (30%) ng·h/ml, 6,009 (42%) ng·h/ml and 15,985 (45%) ng·h/ml in patients with mild, moderate and severe renal impairment. The dosing recommendations in patients with renal impairment, with increased dosing interval, are expected to result in higher peak plasma concentrations and lower C
min levels in patients with renal impairment compared with patients with normal renal function. The clinical implications of this are unknown.
In patients with end-stage renal disease (ESRD) (CrCl < 10 ml/min) requiring haemodialysis, between dialysis tenofovir concentrations substantially increased over 48 hours achieving a mean C
max of 1,032 ng/ml and a mean AUC0-48h of 42,857 ng·h/ml. It is recommended that the dosing interval for tenofovir disoproxil fumarate 300 mg is modified in patients with creatinine clearance < 50 ml/min or in patients who already have ESRD and require dialysis (see Dosage & Administration).
The pharmacokinetics of tenofovir in non-haemodialysis patients with creatinine clearance < 10 ml/min and in patients with ESRD managed by peritoneal or other forms of dialysis have not been studied.
Hepatic impairment: A single 300 mg dose of tenofovir disoproxil fumarate was administered to non-HIV, non-HBV infected patients with varying degrees of hepatic impairment defined according to Child-Pugh-Turcotte (CPT) classification. Tenofovir pharmacokinetic parameters were not substantially altered in subjects with hepatic impairment suggesting that no dose adjustment is required in these subjects. The mean (%CV) tenofovir C
max and AUC0-∞ values were 223 (34.8%) ng/ml and 2,050 (50.8%) ng·h/ml, respectively, in normal subjects compared with 289 (46.0%) ng/ml and 2,31 (43.5%) ng·h/ml in subjects with moderate hepatic impairment, and 305 (24.8%) ng/ml and 2,740 (44.0%) ng·h/ml in subjects with severe hepatic impairment.
Intracellular pharmacokinetics: Tenofovir diphosphate has an intracellular half-life of 10 hours in activated and 50 hours in resting peripheral blood mononuclear cells (PBMCs).
Dolutegravir: Dolutegravir pharmacokinetics are similar between healthy and HIV-infected subjects. The PK variability of dolutegravir is low to moderate. In Phase I studies in healthy subjects, between-subject CVb% for AUC and C
max ranged from ~20 to 40% and C
τ from 30 to 65% across studies. The between-subject PK variability of dolutegravir was higher in HIV-infected subjects than healthy subjects. Within-subject variability (CVw%) is lower than between-subject variability.
Absorption: Dolutegravir is rapidly absorbed following oral administration, with median T
max at 2 to 3 hours post dose for tablet formulation. Food increased the extent and slowed the rate of absorption of dolutegravir. Bioavailability of dolutegravir depends on meal content: low, moderate, and high fat meals increased dolutegravir AUC
(0-∞) by 33%, 41%, and 66%, increased C
max by 46%, 52%, and 67%, prolonged T
max to 3, 4, and 5 hours from 2 hours under fasted conditions, respectively. These increases may be clinically relevant in the presence of certain integrase class resistance. Therefore, Dolutegravir is recommended to be taken with food by patients infected with HIV with integrase class resistance (see Dosage & Administration).
The absolute bioavailability of dolutegravir has not been established.
Distribution: Dolutegravir is highly bound (>99%) to human plasma proteins based on
in vitro data. The apparent volume of distribution is 17 L to 20 L in HIV-infected patients, based on a population pharmacokinetic analysis. Binding of dolutegravir to plasma proteins is independent of dolutegravir concentration. Total blood and plasma drug-related radioactivity concentration ratios averaged between 0.441 to 0.535, indicating minimal association of radioactivity with blood cellular components. The unbound fraction of dolutegravir in plasma is increased at low levels of serum albumin (<35 g/L) as seen in subjects with moderate hepatic impairment.
Dolutegravir is present in cerebrospinal fluid (CSF). In 13 treatment-naïve subjects on a stable dolutegravir plus abacavir/lamivudine regimen, dolutegravir concentration in CSF averaged 18 ng/mL (comparable to unbound plasma concentration, and above the IC
50).
Dolutegravir is present in the female and male genital tract. AUC in cervicovaginal fluid, cervical tissue and vaginal tissue were 6-10% of those in corresponding plasma at steady state. AUC in semen was 7% and 17% in rectal tissue of those in corresponding plasma at steady state.
Biotransformation: Dolutegravir is primarily metabolized through glucuronidation via UGT1A1 with a minor CYP3A component. Dolutegravir is the predominant circulating compound in plasma; renal elimination of unchanged active substance is low (< 1% of the dose). Fifty-three percent of total oral dose is excreted unchanged in the faeces. It is unknown if all or part of this is due to unabsorbed active substance or biliary excretion of the glucuronidate conjugate, which can be further degraded to form the parent compound in the gut lumen. Thirty-two percent of the total oral dose is excreted in the urine, represented by ether glucuronide of dolutegravir (18.9% of total dose), N-dealkylation metabolite (3.6% of total dose), and a metabolite formed by oxidation at the benzylic carbon (3.0% of total dose).
Drug interaction: In vitro, dolutegravir demonstrated no direct, or weak inhibition (IC
50>50
μM) of the enzymes cytochrome P450 (CYP)1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 CYP3A, uridine diphosphate glucuronosyl transferase (UGT)1A1 or UGT2B7, or the transporters Pgp, BCRP, BSEP OATP1B1, OATP1B3, OCT1, MATE2-K, MRP2 or MRP4.
In vitro, dolutegravir did not induce CYP1A2, CYP2B6 or CYP3A4. Based on this data, dolutegravir is not expected to affect the pharmacokinetics of medicinal products that are substrates of major enzymes or transporters (see Interactions).
In vitro, dolutegravir was not a substrate of human OATP 1B1, OATP 1B3 or OCT 1.
Elimination: Dolutegravir has a terminal half-life of ~14 hours. The apparent oral clearance (CL/F) is approximately 1L/hr in HIV-infected patients based on a population pharmacokinetic analysis.
Linearity/non-linearity: The linearity of dolutegravir pharmacokinetics is dependent on dose and formulation. Following oral administration of tablet formulations, in general, dolutegravir exhibited nonlinear pharmacokinetics with less than dose-proportional increases in plasma exposure from 2 to 100 mg; however increase in dolutegravir exposure appears dose proportional from 25 mg to 50 mg for the tablet formulation. With 50 mg twice daily, the exposure over 24 hours was approximately doubled compared to 50 mg once daily.
Pharmacokinetic/pharmacodynamic relationship(s): In a randomized, dose-ranging trial, HIV-1-infected subjects treated with dolutegravir monotherapy (ING111521) demonstrated rapid and dose-dependent antiviral activity, with mean decline in HIV-1 RNA of 2.5 log10 at day 11 for 50 mg dose. This antiviral response was maintained for 3 to 4 days after the last dose in the 50 mg group.
PK/PD modelling using pooled data from clinical studies in integrase resistant patients suggest that increasing the dose from 50 mg twice daily to 100 mg twice daily may increase the effectiveness of dolutegravir in patients with integrase resistance and limited treatment options due to advanced multi class resistance. The proportion of responders (HIV-1 RNA <50 c/mL) at week 24 was predicted to increase around 4-18% in the subjects with Q148 + ≥2 secondary mutations from G140A/C/S, E138A/K/T, L74I. Although these simulated results have not been confirmed in clinical trials, this high dose may be considered in the presence of the Q148 + ≥2 secondary mutations from G140A/C/S, E138A/K/T, L74I in patients with overall limited treatment options due to advanced multi class resistance. There is no clinical data on the safety or efficacy of the 100 mg twice daily dose. Co-treatment with atazanavir increases the exposure of dolutegravir markedly, and should not be used in combination with this high dose, since safety with the resulting dolutegravir exposure has not been established.
Special patient populations: Children:
The pharmacokinetics of dolutegravir in 10 antiretroviral treatment-experienced HIV-1 infected adolescents (12 to <18 years of age) showed that Dolutegravir 50 mg once daily oral dosage resulted in dolutegravir exposure comparable to that observed in adults who received Dolutegravir 50 mg orally once daily.
Elderly: Population pharmacokinetic analysis of dolutegravir using data in HIV-1 infected adults showed that there was no clinically relevant effect of age on dolutegravir exposure. Pharmacokinetic data for dolutegravir in subjects >65 years of age are limited.
Renal impairment: Renal clearance of unchanged active substance is a minor pathway of elimination for dolutegravir. A study of the pharmacokinetics of dolutegravir was performed in subjects with severe renal impairment (CLcr <30 mL/min) and matched healthy controls. The exposure to dolutegravir was decreased by approximately 40% in subjects with severe renal impairment. The mechanism for the decrease is unknown. No dosage adjustment is considered necessary for patients with renal impairment. Dolutegravir has not been studied in patients on dialysis.
Hepatic impairment: Dolutegravir is primarily metabolized and eliminated by the liver. A single dose of 50 mg of dolutegravir was administered to 8 subjects with moderate hepatic impairment (Child-Pugh class B) and to 8 matched healthy adult controls. While the total dolutegravir concentration in plasma was similar, a 1.5- to 2-fold increase in unbound exposure to dolutegravir was observed in subjects with moderate hepatic impairment compared to healthy controls. No dosage adjustment is considered necessary for patients with mild to moderate hepatic impairment. The effect of severe hepatic impairment on the pharmacokinetics of Dolutegravir has not been studied.
Polymorphisms in drug metabolising enzymes: There is no evidence that common polymorphisms in drug metabolising enzymes alter dolutegravir pharmacokinetics to a clinically meaningful extent. In a meta-analysis using pharmacogenomics samples collected in clinical studies in healthy subjects, subjects with UGT1A1 (n=7) genotypes conferring poor dolutegravir metabolism had a 32% lower clearance of dolutegravir and 46% higher AUC compared with subjects with genotypes associated with normal metabolism via UGT1A1 (n=41).
Gender: Population PK analyses using pooled pharmacokinetic data from Phase IIb and Phase III adult trials revealed no clinically relevant effect of gender on the exposure of dolutegravir.
Race: Population PK analyses using pooled pharmacokinetic data from Phase IIb and Phase III adult trials revealed no clinically relevant effect of race on the exposure of dolutegravir. The pharmacokinetics of dolutegravir following single dose oral administration to Japanese subjects appear similar to observed parameters in Western (US) subjects.
Co-infection with Hepatitis B or C: Population pharmacokinetic analysis indicated that hepatitis C virus co-infection had no clinically relevant effect on the exposure to dolutegravir. There are limited data on subjects with hepatitis B co-infection.
Toxicology: Preclinical Safety Data: Administration of lamivudine in animal toxicity studies at high doses was not associated with any major organ toxicity. Lamivudine was not mutagenic in bacterial tests but showed activity in an
in vitro cytogenetic assay and the mouse lymphoma assay. Lamivudine was not genotoxic
in vitro at doses that gave plasma concentrations around 40-50 times higher than the anticipated clinical plasma levels. As the
in vitro mutagenic activity of lamivudine could not be confirmed in
in vivo tests, it is concluded that lamivudine should not represent a genotoxic hazard to patients undergoing treatment.
The results of long-term carcinogenicity studies in rats and mice did not show any carcinogenic potential relevant for humans.
Tenofovir: Preclinical studies conducted in rats, dogs and monkeys revealed target organ effects in gastrointestinal tract, kidney, bone and a decrease in serum phosphate concentration. Bone toxicity was diagnosed as osteomalacia (monkeys) and reduced bone mineral density (rats and dogs). Findings in the rat and monkey studies indicated that there was a substance-related decrease in intestinal absorption of phosphate with potential secondary reduction in bone mineral density. However, no conclusion could be drawn on the mechanism(s) underlying these toxicities.
Reproductive studies were conducted in rats and rabbits. There were no effects on mating or fertility parameters or on any pregnancy or foetal parameter. There were no gross foetal alterations of soft or skeletal tissues. Tenofovir disoproxil fumarate reduced the viability index and weight of pups in peri-post natal toxicity studies.
Genotoxicity studies have shown that tenofovir disoproxil fumarate was negative in the
in vivo mouse bone marrow micronucleus assay but was positive for inducing forward mutations in the
in vitro L5178Y mouse lymphoma cell assay in the presence or absence of S9 metabolic activation. Tenofovir disoproxil fumarate was positive in the Ames test (strain TA 1535) in two out of three studies, once in the presence of S9 mix (6.2- to 6.8-fold increase) and once without S9 mix. Tenofovir disoproxil fumarate was also weakly positive in an
in vivo/in vitro unscheduled DNA synthesis test in primary rat hepatocytes.
Tenofovir disoproxil fumarate did not show any carcinogenic potential in a long-term oral carcinogenicity study in rats. A long-term oral carcinogenicity study in mice showed a low incidence of duodenal tumours, considered likely related to high local concentrations of tenofovir disoproxil fumarate in the gastrointestinal tract at a dose of 600 mg/kg/day. While the mechanism of tumour formation is uncertain, the findings are unlikely to be of relevance to humans.
Dolutegravir: Dolutegravir was not mutagenic or clastogenic using in vitro tests in bacteria and cultured mammalian cells, and an
in vivo rodent micronucleus assay. Dolutegravir was not carcinogenic in long term studies in the mouse and rat.
Dolutegravir did not affect male or female fertility in rats at doses up to 1000 mg/kg/day, the highest dose tested (24 times the 50 mg twice daily human clinical exposure based on AUC).
Oral administration of dolutegravir to pregnant rats at doses up to 1000 mg/kg daily from days 6 to 17 of gestation did not elicit maternal toxicity, developmental toxicity or teratogenicity (27 times the 50 mg twice daily human clinical exposure based on AUC).
Oral administration of dolutegravir to pregnant rabbits at doses up to 1000 mg/kg daily from days 6 to 18 of gestation did not elicit developmental toxicity or teratogenicity (0.40 times the 50 mg twice daily human clinical exposure based on AUC). In rabbits, maternal toxicity (decreased food consumption, scant/no faeces/urine, suppressed body weight gain) was observed at 1000 mg/kg (0.40 times the 50 mg twice daily human clinical exposure based on AUC).
The effect of prolonged daily treatment with high doses of dolutegravir has been evaluated in repeat oral dose toxicity studies in rats (up to 26 weeks) and in monkeys (up to 38 weeks). The primary effect of dolutegravir was gastrointestinal intolerance or irritation in rats and monkeys at doses that produce systemic exposures approximately 21 and 0.82 times the 50 mg twice daily human clinical exposure based on AUC, respectively. Because gastrointestinal (GI) intolerance is considered to be due to local active substance administration, mg/kg or mg/m
2 metrics are appropriate determinates of safety cover for this toxicity. GI intolerance in monkeys occurred at 15 times the human mg/kg equivalent dose (based on a 50 kg human), and 5 times the human mg/m
2 equivalent dose for a clinical dose of 50 mg twice daily.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image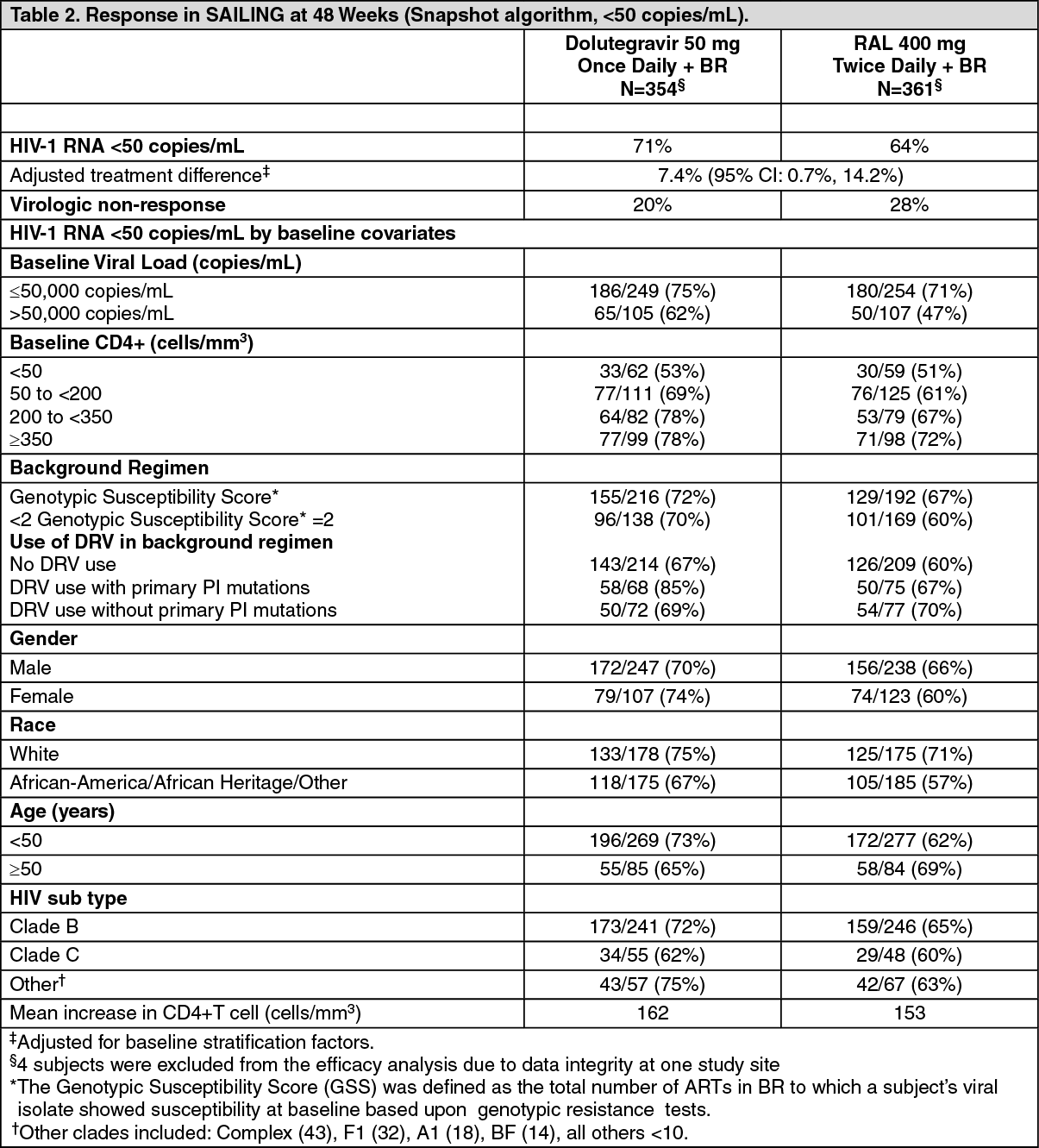 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image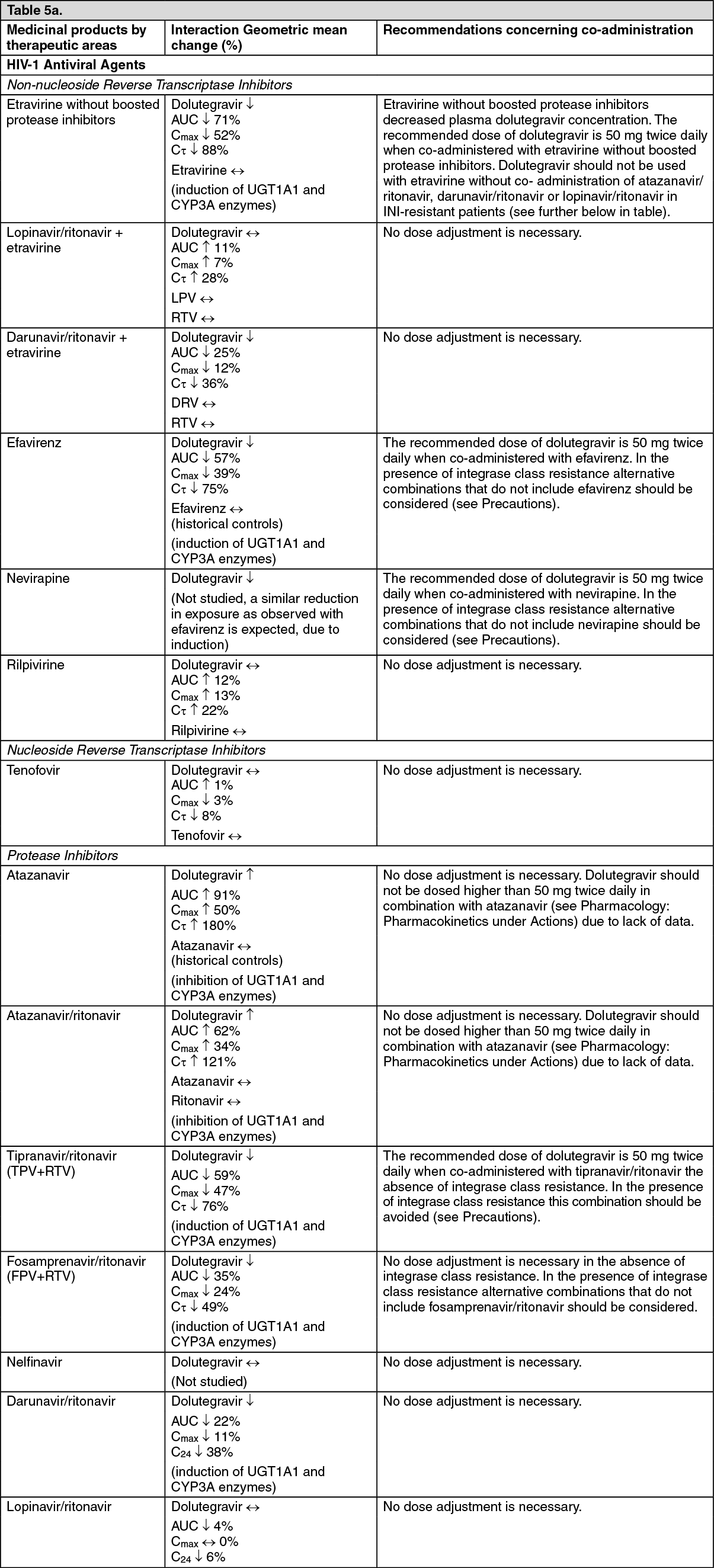 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image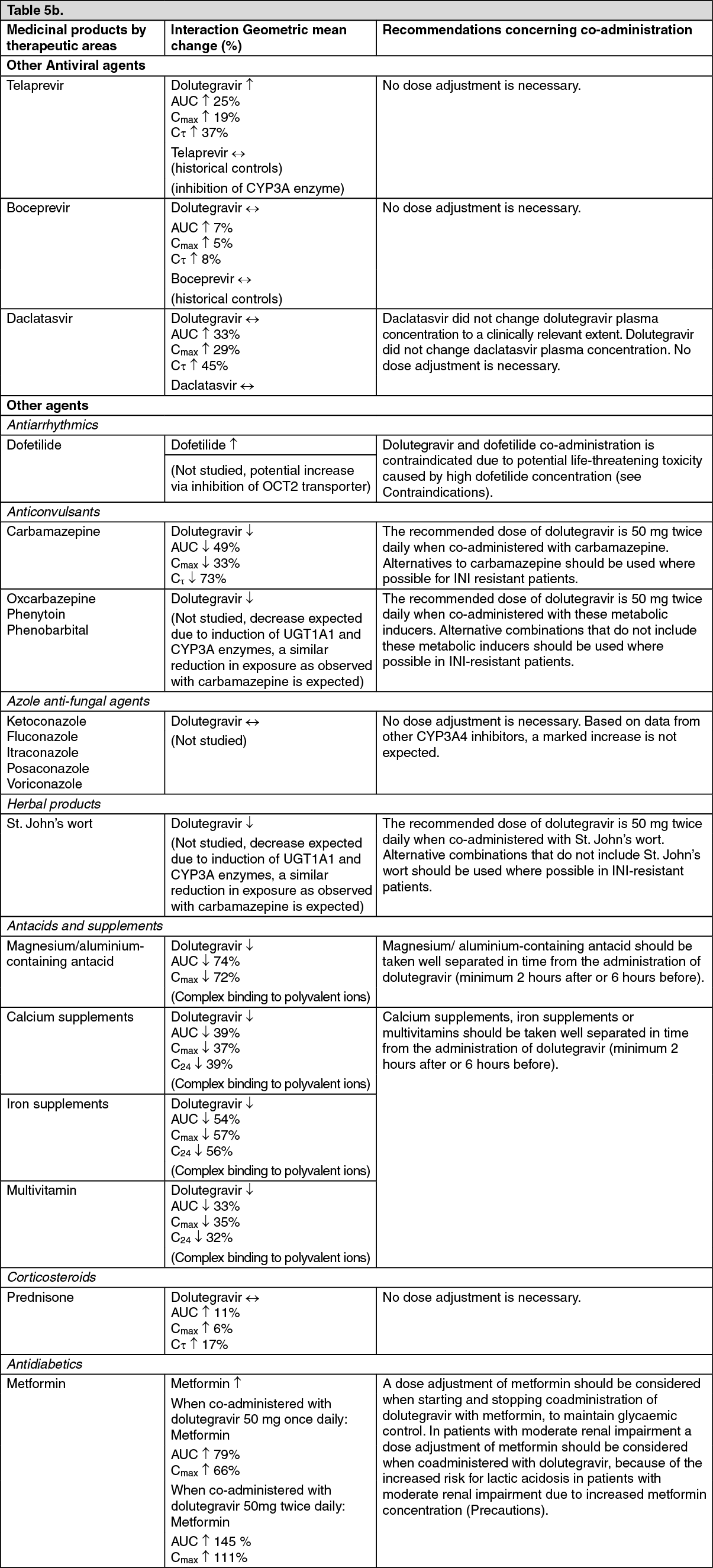 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image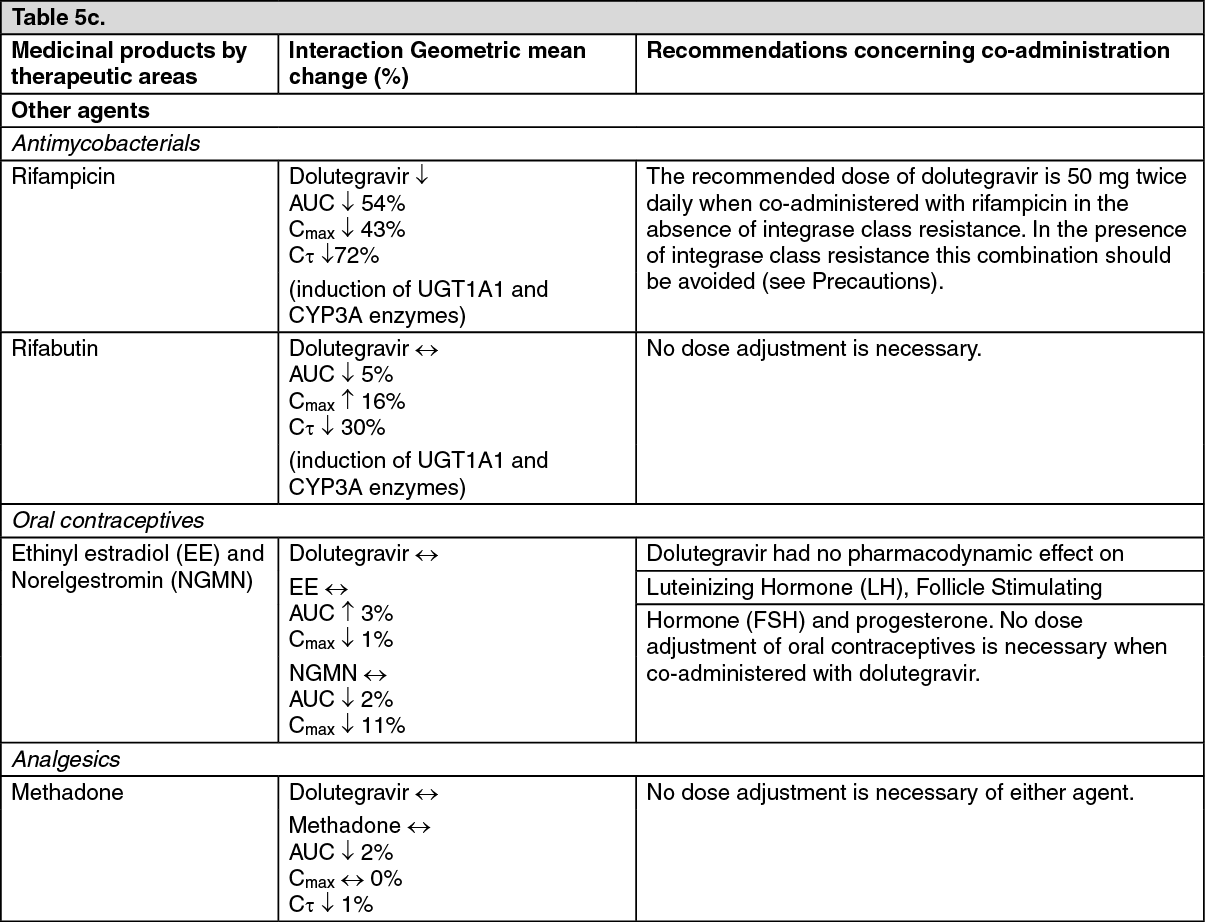 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out