Therapy should be initiated by a physician experienced in the treatment of patients with haematological malignancies as appropriate.
For doses of 400 mg and above (see dosage recommendation as follows) a 400 mg tablet (not divisible) is available.
The prescribed dose should be administered orally with a meal and a large glass of water to minimise the risk of gastrointestinal irritations. Doses of 400 mg or 600 mg should be administered once daily, whereas a daily dose of 800 mg should be administered as 400 mg twice a day, in the morning and in the evening.
For patients unable to swallow the film-coated tablets, the tablets may be dispersed in a glass of mineral water or apple juice. The required number of tablets should be placed in the appropriate volume of beverage (approximately 50 ml for a 100 mg tablet, and 200 ml for a 400 mg tablet) and stirred with a spoon. The suspension should be administered immediately after complete disintegration of the tablet(s).
Posology for CML in adult patients: The recommended dose of Alvotinib is 600 mg/day for patients in blast crisis. Blast crisis is defined as blasts ≥30% in blood or bone marrow or extramedullary disease other than hepatosplenomegaly.
Treatment duration: The effect of stopping treatment after the achievement of a complete cytogenetic response has not been investigated.
Dose increases from 600 mg to a maximum of 800 mg (given as 400 mg twice daily) in patients with blast crisis may be considered in the absence of severe adverse drug reaction and severe non-leukaemia-related neutropenia or thrombocytopenia in the following circumstances: failure to achieve a satisfactory haematological response after at least 3 months of treatment; failure to achieve a cytogenetic response after 12 months of treatment; or loss of a previously achieved haematological and/or cytogenetic response. Patients should be monitored closely following dose escalation given the potential for an increased incidence of adverse reactions at higher dosages.
Posology for CML in children: Dosing in children should be on the basis of body surface area (mg/m2). The dose of 340 mg/m2 daily is recommended for children with chronic phase CML and advanced phase CML (not to exceed the total dose of 600 mg daily). Treatment can be given as a once daily dose or alternatively the daily dose may be split into two administrations - one in the morning and one in the evening.
The dose recommendation is currently based on a small number of paediatric patients.
There is no experience with the use of Alvotinib in children below 2 years of age.
Dosage in GIST: The recommended dose of Alvotinib is 400 mg/day for patients with unresectable and/or metastatic, malignant GIST.
A dose increase from 400 mg to 600 mg or 800 mg for patients may be considered in the absence of adverse drug reactions if assessments demonstrate an insufficient response to therapy.
Treatment with Alvotinib in GIST patients should be continued until disease progression.
The recommended dose of Alvotinib is 400 mg/day for the adjuvant treatment of adult patients following resection of GIST. In the adjuvant setting the optimal treatment duration with Alvotinib is not known.
Dose adjustment for adverse reactions:
Non-haematological adverse reactions:
If a severe non-haematological adverse reaction develops with Alvotinib use, treatment must be withheld until the event has resolved. Thereafter, treatment can be resumed as appropriate depending on the initial severity of the event.
If elevations in bilirubin >3 x institutional upper limit of normal (IULN) or in liver transaminases >5 x IULN occur, Alvotinib should be withheld until bilirubin levels have returned to <1.5 x IULN and transaminase levels to <2.5 x IULN. Treatment with Alvotinib may then be continued at a reduced daily dose. In adults the dose should be reduced from 600 to 400 mg, or from 800 mg to 600 mg, and in children from 340 to 260 mg/m
2/day.
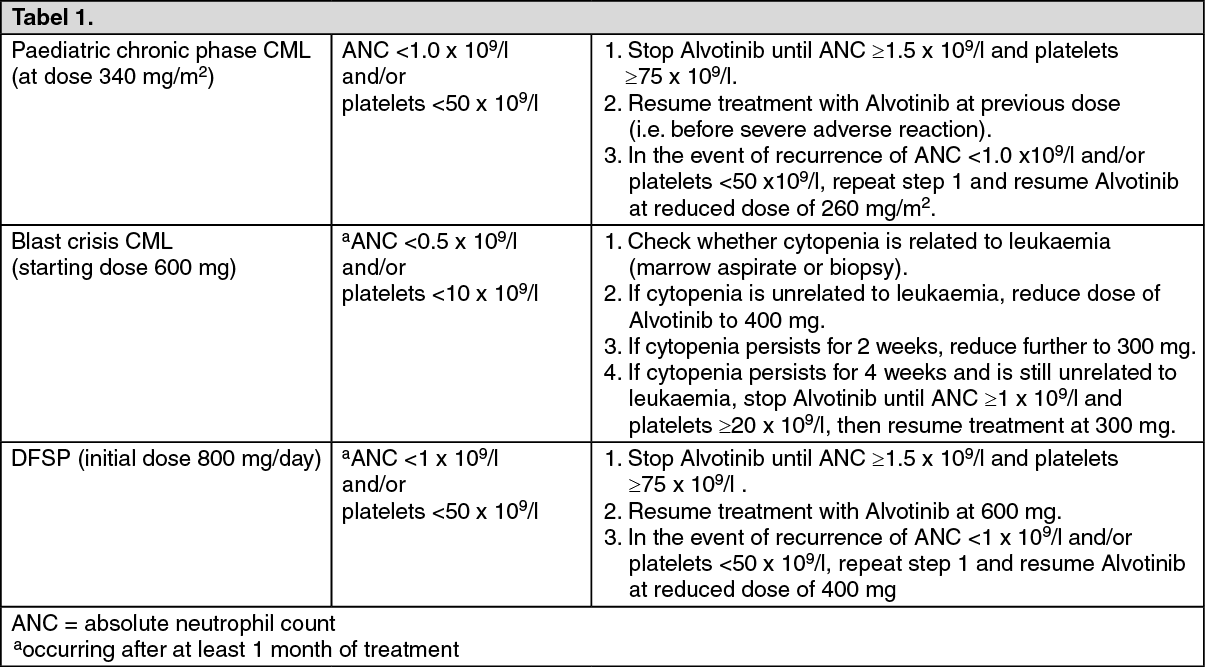
Haematological adverse reactions: Dose reduction or treatment interruption for severe neutropenia and thrombocytopenia are recommended as indicated in Table 1.
Dose adjustments for neutropenia and thrombocytopenia: See Table 1.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Paediatric use: There is no experience in children with CML below 2 years of age (see Pharmacology: Pharmacodynamics under Actions).
Hepatic insufficiency: Imatinib is mainly metabolised through the liver. Patients with mild, moderate or severe liver dysfunction should be given the minimum recommended dose of 400 mg daily. The dose can be reduced if not tolerated (see Precautions, Adverse Reactions and Pharmacology: Pharmacokinetics under Actions).
Liver dysfunction classification: See Table 2.
Click on icon to see table/diagram/image


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image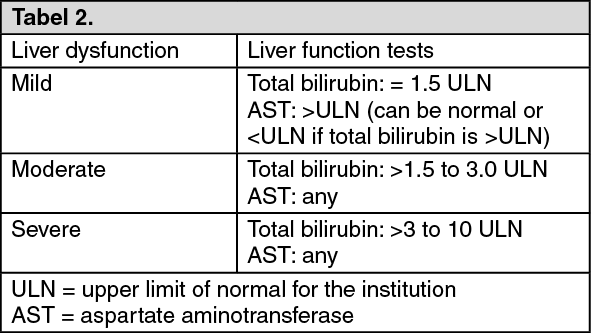 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
