Pharmacotherapeutic group: Antineoplastic and immunomodulating agents, antineoplastic agents, other antineoplastic agents, monoclonal antibodies.
ATC code: L01X C07.
Pharmacology: Pharmacodynamics: Mechanism of action: Bevacizumab binds to vascular endothelial growth factor (VEGF), the key driver of vasculogenesis and angiogenesis, and thereby inhibits the binding of VEGF to its receptors, Flt-1 (VEGFR-1) and KDR (VEGFR-2), on the surface of endothelial cells. Neutralizing the biological activity of VEGF regresses the vascularisation of tumours, normalises remaining tumour vasculature, and inhibits the formation of new tumour vasculature, thereby inhibiting tumour growth.
Pharmacodynamic effects: Administration of bevacizumab or its parental murine antibody to xenotransplant models of cancer in nude mice resulted in extensive anti-tumour activity in human cancers, including colon, breast, pancreas and prostate. Metastatic disease progression was inhibited and microvascular permeability was reduced.
Clinical efficacy: The information about the clinical efficacy of bevacizumab is presented according to review of literature data.
Metastatic carcinoma of the colon or rectum (mCRC): The safety and efficacy of the recommended dose (5 mg/kg of body weight every two weeks) in metastatic carcinoma of the colon or rectum were studied in three randomised, active-controlled clinical trials in combination with fluoropyrimidine-based first-line chemotherapy. Bevacizumab was combined with two chemotherapy regimens: AVF2107g: A weekly schedule of irinotecan/bolus 5-fluorouracil/folinic acid (IFL) for a total of 4 weeks of each 6 week-cycle (Saltz regimen).
AVF0780g: In combination with bolus 5-fluorouracil/folinic acid (5-FU/FA) for a total of 6 weeks of each 8 week-cycle (Roswell Park regimen).
AVF2192g: In combination with bolus 5-FU/FA for a total of 6 weeks of each 8 week-cycle (Roswell Park regimen) in patients who were not optimal candidates for first-line irinotecan treatment.
Three additional studies with bevacizumab have been conducted in mCRC patients: first-line (NO16966), second-line with no previous bevacizumab treatment (E3200), and second-line with previous bevacizumab treatment following disease progression in first-line (ML18147). In these studies, bevacizumab was administered at the following dosing regimens in combination with FOLFOX-4 (5-FU/LV/oxaliplatin), XELOX (capecitabine/oxaliplatin), and fluoropyrimidine/irinotecan and fluoropyrimidine/oxaliplatin: NO16966: Bevacizumab 7.5 mg/kg of body weight every 3 weeks in combination with oral capecitabine and intravenous oxaliplatin (XELOX) or bevacizumab 5 mg/kg every 2 weeks in combination with leucovorin plus 5-fluorouracil bolus, followed by 5-fluorouracil infusion, with intravenous oxaliplatin (FOLFOX-4).
E3200: Bevacizumab 10 mg/kg of body weight every 2 weeks in combination with leucovorin and 5-fluorouracil bolus, followed by 5-fluorouracil infusion, with intravenous oxaliplatin (FOLFOX-4) in bevacizumab-naïve patients.
ML18147: Bevacizumab 5.0 mg/kg of body weight every 2 weeks or bevacizumab 7.5 mg/kg of body weight every 3 weeks in combination with fluoropyrimidine/irinotecan or fluoropyrimidine/oxaliplatin in patients with disease progression following first-line treatment with bevacizumab. Use of irinotecan- or oxaliplatin-containing regimen was switched depending on first-line usage of either oxaliplatin or irinotecan.
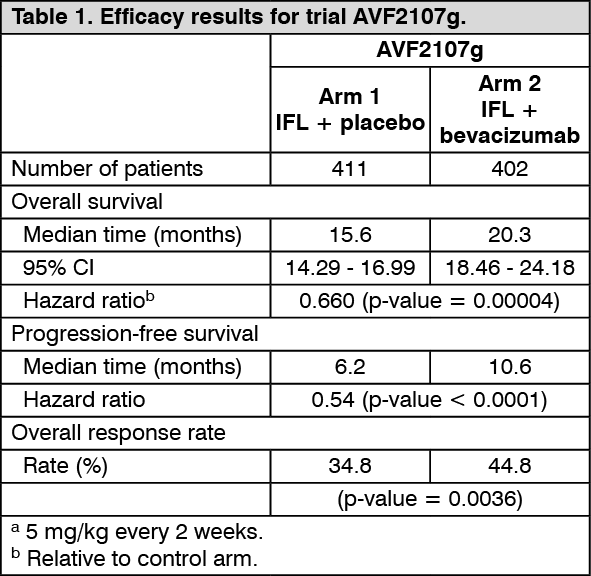
AVF2107g: This was a phase III randomised, double-blind, active-controlled clinical trial evaluating bevacizumab in combination with IFL as first-line treatment for metastatic carcinoma of the colon or rectum. Eight hundred and thirteen patients were randomised to receive IFL + placebo (Arm 1) or IFL + bevacizumab (5 mg/kg every 2 weeks, Arm 2). A third group of 110 patients received bolus 5-FU/FA + bevacizumab (Arm 3). Enrolment in Arm 3 was discontinued, as pre-specified, once safety of bevacizumab with the IFL regimen was established and considered acceptable. All treatments were continued until disease progression. The overall mean age was 59.4 years; 56.6% of patients had an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0, 43% had a value of 1 and 0.4% had a value of 2. 15.5% of patients received prior radiotherapy and 28.4% underwent prior chemotherapy.
The primary efficacy variable of the trial was overall survival. The addition of bevacizumab to IFL resulted in statistically significant increases in overall survival, progression-free survival and overall response rate (see Table 1). The clinical benefit, as measured by overall survival, was seen in all pre-specified patient subgroups, including those defined by age, sex, PS, location of primary tumour, number of organs involved and duration of metastatic disease.
The efficacy results of bevacizumab in combination with IFL-chemotherapy are displayed in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Among the 110 patients randomised to Arm 3 (5-FU/FA + bevacizumab) prior to discontinuation of this arm, the median overall survival was 18.3 months and the median progression free survival was 8.8 months.
AVF2192g: This was a phase II randomised, double-blind, active-controlled clinical trial evaluating the efficacy and safety of bevacizumab in combination with 5-FU/FA as first-line treatment for metastatic colorectal cancer in patients who were not optimal candidates for first-line irinotecan treatment. One hundred and five patients were randomised to 5-FU/FA + placebo arm and 104 patients to 5-FU/FA + bevacizumab (5 mg/kg every 2 weeks) arm. All treatments were continued until disease progression. The addition of bevacizumab 5 mg/kg every two weeks to 5-FU/FA resulted in higher objective response rates (ORR), significantly longer progression-free survival, and a trend in longer survival as compared to 5-FU/FA chemotherapy alone.
AVF0780g: This was a phase II randomised, active-controlled, open-labelled clinical trial investigating bevacizumab in combination with 5-FU/FA as first-line treatment of metastatic colorectal cancer. The median age was 64 years. 19% of the patients underwent prior chemotherapy and 14% had received prior radiotherapy.
Seventy-one patients were randomised to receive bolus 5-FU/FA or 5-FU/FA + bevacizumab (5 mg/kg every 2 weeks). A third group of 33 patients received bolus 5-FU/FA + bevacizumab (10 mg/kg every 2 weeks). Patients were treated until disease progression. The primary endpoints of the trial were objective response rate and progression-free survival. The addition of bevacizumab 5 mg/kg every two weeks to 5-FU/FA resulted in higher objective response rates, longer progression-free survival, and a trend in longer survival, compared with 5-FU/FA chemotherapy alone (see Table 2). These efficacy data are consistent with the results from trial AVF2107g.
The efficacy data from trials AVF0780g and AVF2192g investigating bevacizumab in combination with 5-FU/FA-chemotherapy are summarized in Table 2. (See Table 2.)
Click on icon to see table/diagram/image
NO16966: This was a phase III randomised, double-blind (for bevacizumab), clinical trial investigating bevacizumab 7.5 mg/kg in combination with oral capecitabine and IV oxaliplatin (XELOX), administered on a 3-weekly schedule; or bevacizumab 5 mg/kg in combination with leucovorin with 5-fluorouracil bolus, followed by 5-fluorouracil infusional, with IV oxaliplatin (FOLFOX-4), administered on a 2-weekly schedule. The trial contained two parts: an initial unblinded 2-arm part (Part I) in which patients were randomised to two different treatment groups (XELOX and FOLFOX-4) and a subsequent 2 x 2 factorial 4-arm part (Part II) in which patients were randomised to four treatment groups (XELOX + placebo, FOLFOX-4 + placebo, XELOX + bevacizumab, FOLFOX-4 + bevacizumab). In Part II, treatment assignment was double-blind with respect to bevacizumab.
Approximately 350 patients were randomised into each of the 4 trial arms in the Part II of the trial. (See Table 3.)
Click on icon to see table/diagram/image
The primary efficacy parameter of the trial was the duration of progression-free survival. In this trial, there were two primary objectives: to show that XELOX was non-inferior to FOLFOX-4 and to show that bevacizumab in combination with FOLFOX-4 or XELOX chemotherapy was superior to chemotherapy alone. Both co-primary objectives were met: Non-inferiority of the XELOX-containing arms compared with the FOLFOX-4-containing arms in the overall comparison was demonstrated in terms of progression-free survival and overall survival in the eligible per-protocol population.
Superiority of the bevacizumab-containing arms versus the chemotherapy alone arms in the overall comparison was demonstrated in terms of progression-free survival in the ITT population (Table 4).
Secondary PFS analyses, based on 'on-treatment'-based response assessments, confirmed the significantly superior clinical benefit for patients treated with bevacizumab (analyses shown in Table 4), consistent with the statistically significant benefit observed in the pooled analysis. (See Table 4.)
Click on icon to see table/diagram/image
In the FOLFOX treatment subgroup, the median PFS was 8.6 months in placebo and 9.4 months in bevacizumab treated patients, HR = 0.89, 97.5% CI = [0.73; 1.08]; p-value = 0.1871, the corresponding results in the XELOX treatment subgroup being 7.4 vs. 9.3 months, HR = 0.77, 97.5% CI = [0.63; 0.94]; p-value = 0.0026.
The median overall survival was 20.3 months in placebo and 21.2 months in bevacizumab treated patients in the FOLFOX treatment subgroup, HR=0.94, 97.5% CI = [0.75; 1.16]; p-value = 0.4937, the corresponding results in the XELOX, treatment subgroup being 19.2 vs. 21.4 months, HR = 0.84, 97.5% CI = [0.68; 1.04]; p-value = 0.0698.
ECOG E3200: This was a phase III randomised, active-controlled, open-label trial investigating bevacizumab 10 mg/kg in combination with leucovorin with 5-fluorouracil bolus and then 5-fluorouracil IV, with IV oxaliplatin (FOLFOX-4), administered on a 2-weekly schedule in previously-treated patients (second line) with advanced colorectal cancer. In the chemotherapy arms, the FOLFOX-4 regimen used the same doses and schedule as shown in Table 3 for trial NO16966.
The primary efficacy parameter of the trial was overall survival, defined as the time from randomisation to death from any cause. Eight hundred and twenty-nine patients were randomised (292 FOLFOX-4, 293 bevacizumab + FOLFOX-4 and 244 bevacizumab monotherapy). The addition of bevacizumab to FOLFOX-4 resulted in a statistically significant prolongation of survival. Statistically significant improvements in progression-free survival and objective response rate were also observed (see Table 5).
Click on icon to see table/diagram/image
No significant difference was observed in the duration of overall survival between patients who received bevacizumab monotherapy compared to patients treated with FOLFOX-4. Progression-free survival and objective response rate were inferior in the bevacizumab monotherapy arm compared to the FOLFOX-4 arm.
ML18147: This was a Phase III randomised, controlled, open-label trial investigating bevacizumab 5.0 mg/kg every 2 weeks or 7.5 mg/kg every 3 weeks in combination with fluoropyrimidine-based chemotherapy versus fluoropyrimidine-based chemotherapy alone in patients with mCRC who have progressed on a first-line bevacizumab-containing regimen.
Patients with histologically confirmed mCRC and disease progression were randomised 1:1 within 3 months after discontinuation of bevacizumab first-line therapy to receive fluoropyrimidine/oxaliplatin- or fluoropyrimidine/irinotecan-based chemotherapy (chemotherapy switched depending on first-line chemotherapy) with or without bevacizumab. Treatment was given until progressive disease or unacceptable toxicity. The primary end point was overall survival defined as the time from randomisation until death from any cause.
A total of 820 patients were randomised. The addition of bevacizumab to fluoropyrimidine-based chemotherapy resulted in a statistically significant prolongation of survival in patients with mCRC who have progressed on a first-line bevacizumab-containing regimen (ITT = 819) (see Table 6).
Click on icon to see table/diagram/image
Statistically significant improvements in progression-free survival were also observed. Objective response rate was low in both treatment arms and the difference was not significant.
Study E3200 used a 5 mg/kg/week equivalent dose of bevacizumab in bevacizumab-naïve patients, while study ML18147 used a 2.5 mg/kg/week equivalent dose of bevacizumab in bevacizumab-pretreated patients. A cross-trial comparison of the efficacy and safety data is limited by differences between these studies, most notably in patient populations, previous bevacizumab exposure and chemotherapy regimens. Both the 5 mg/kg/week and 2.5 mg/kg/week equivalent doses of bevacizumab provided a statistically significant benefit with regards to OS (HR 0.751 in study E3200; HR 0.81 in study ML18147) and PFS (HR 0.518 in study E3200; HR 0.68 in study ML18147). In terms of safety, there was a higher overall incidence of Grade 3-5 AEs in study E3200 relative to study ML18147.
Metastatic breast cancer (mBC): Two large Phase III trials were designed to investigate the treatment effect of bevacizumab in combination with two individual chemotherapy agents, as measured by the primary endpoint of PFS. A clinically meaningful and statistically significant improvement in PFS was observed in both trials.
Summarised as follows are PFS results for the individual chemotherapy agents included in the indication: Study E2100 (paclitaxel): Median PFS increase 5.6 months, HR 0.421 (p < 0.0001, 95% CI 0.343; 0.516).
Study AVF3694g (capecitabine): Median PFS increase 2.9 months, HR 0.69 (p = 0.0002, 95% CI 0.56; 0.84).
Further details of each study and the results are provided as follows.
ECOG E2100: Trial E2100 was an open-label, randomised, active controlled, multicentre clinical trial evaluating bevacizumab in combination with paclitaxel for locally recurrent or metastatic breast cancer in patients who had not previously received chemotherapy for locally recurrent and metastatic disease. Patients were randomised to paclitaxel alone (90 mg/m
2 IV over 1 hour once weekly for three out of four weeks) or in combination with bevacizumab (10 mg/kg IV infusion every two weeks). Prior hormone therapy for the treatment of metastatic disease was allowed. Adjuvant taxane therapy was allowed only if it was completed at least 12 months prior to trial entry. Of the 722 patients in the trial, the majority of patients had HER2-negative disease (90%), with a small number of patients with unknown (8%) or confirmed HER2-positive status (2%), who had previously been treated with or were considered unsuitable for trastuzumab therapy. Furthermore, 65% of patients had received adjuvant chemotherapy including 19% prior taxanes and 49% prior anthracyclines. Patients with central nervous system metastases, including previously treated or resected brain lesions, were excluded.
In trial E2100, patients were treated until disease progression. In situations where early discontinuation of chemotherapy was required, treatment with bevacizumab as a single agent continued until disease progression. Patient characteristics were similar across the trial arms. The primary endpoint of this trial was progression free survival (PFS), based on trial investigators' assessment of disease progression. In addition, an independent review of the primary endpoint was also conducted. The results of this trial are presented in Table 7. (See Table 7.)
Click on icon to see table/diagram/image
The clinical benefit of bevacizumab as measured by PFS was seen in all pre-specified subgroups tested (including disease-free interval, number of metastatic sites, prior receipt of adjuvant chemotherapy and oestrogen receptor (ER) status).
AVF3694g: Study AVF3694g was a Phase III, multicentre, randomised, placebo-controlled trial designed to evaluate the efficacy and safety of bevacizumab in combination with chemotherapy compared to chemotherapy plus placebo as first-line treatment for patients with HER2-negative metastatic or locally recurrent breast cancer.
Chemotherapy was chosen at the investigator's discretion prior to randomisation in a 2:1 ratio to receive either chemotherapy plus bevacizumab or chemotherapy plus placebo. The choices of chemotherapy included capecitabine, taxane (protein-bound paclitaxel, docetaxel), and anthracycline-based agents (doxorubicin/cyclophosphamide, epirubicin/cyclophosphamide, 5-fluorouracil/doxorubicin/cyclophosphamide, 5-fluorouracil/epirubicin/cyclophosphamide) given every three weeks (q3w). Bevacizumab or placebo was administered at a dose of 15 mg/kg q3w. This study included a blinded treatment phase, an optional open-label post-progression phase, and a survival follow-up phase. During the blinded treatment phase, patients received chemotherapy and medicinal product (bevacizumab or placebo) every 3 weeks until disease progression, treatment-limiting toxicity, or death. On documented disease progression, patients who entered the optional open-label phase could receive open-label bevacizumab together with a wide-range of second line therapies.
Statistical analyses were performed independently for 1) patients who received capecitabine in combination with bevacizumab or placebo; 2) patients who received taxane-based or anthracycline-based chemotherapy in combination with bevacizumab or placebo. The primary endpoint of the study was PFS by investigator assessment. In addition, the primary endpoint was also assessed by an independent review committee (IRC).
The results of this study from the final protocol defined analyses for PFS and response rates for the independently powered capecitabine cohort of Study AVF3694g are presented in Table 8 Results from an exploratory overall survival analysis which include an additional 7 months of follow-up (approximately 46% of patients had died) are also presented. The percentage of patients who received bevacizumab in the open-label phase was 62.1% in the capecitabine + placebo arm and 49.9% in the capecitabine + bevacizumab arm. (See Table 8.)
Click on icon to see table/diagram/image
An unstratified analysis of PFS (investigator assessed) was performed that did not censor for non-protocol therapy prior to disease progression. The results of these analyses were very similar to the primary PFS results.
Non-small cell lung cancer (NSCLC): First-line treatment of non-squamous NSCLC in combination with platinum-based chemotherapy: The safety and efficacy of bevacizumab, in addition to platinum-based chemotherapy, in the first-line treatment of patients with non-squamous NSCLC, was investigated in trials E4599 and BO17704. An overall survival benefit has been demonstrated in trial E4599 with a 15 mg/kg/q3wk dose of bevacizumab. Trial BO17704 has demonstrated that both 7.5 mg/kg/q3wk and 15 mg/kg/q3wk bevacizumab doses increase PFS and response rate.
E4599: E4599 was an open-label, randomised, active-controlled, multicentre clinical trial evaluating bevacizumab as first-line treatment of patients with locally advanced (stage IIIb with malignant pleural effusion), metastatic or recurrent NSCLC other than predominantly squamous cell histology.
Patients were randomised to platinum-based chemotherapy (paclitaxel 200 mg/m
2) and carboplatin AUC = 6.0, both by IV infusion (PC) on day 1 of every 3-week cycle for up to 6 cycles or PC in combination with bevacizumab at a dose of 15 mg/kg IV infusion day 1 of every 3-week cycle. After completion of six cycles of carboplatin-paclitaxel chemotherapy or upon premature discontinuation of chemotherapy, patients on the bevacizumab + carboplatin-paclitaxel arm continued to receive bevacizumab as a single agent every 3 weeks until disease progression. 878 patients were randomised to the two arms.
During the trial, of patients who received trial treatment, 32.2% (136/422) of patients received 7-12 administrations of bevacizumab and 21.1% (89/422) of patients received 13 or more administrations of bevacizumab.
The primary endpoint was overall survival. Results are presented in Table 9. (See Table 9.)
Click on icon to see table/diagram/image
In an exploratory analysis, the extent of bevacizumab benefit on overall survival was less pronounced in non-adenocarcinoma tumor subgroup.
BO17704: Trial BO17704 was a randomised, double-blind phase III trial of bevacizumab in addition to cisplatin and gemcitabine versus placebo, cisplatin and gemcitabine in patients with locally advanced (stage IIIb with supraclavicular lymph node metastases or with malignant pleural or pericardial effusion), metastatic or recurrent non-squamous NSCLC, who had not received prior chemotherapy. The primary endpoint was PFS, secondary endpoints for the trial included the duration of overall survival.
Patients were randomized to platinum-based chemotherapy, cisplatin 80 mg/m
2 intravenous infusion on day 1 and gemcitabine 1250 mg/m
2 intravenous infusion on days 1 and 8 of every 3-week cycle for up to 6 cycles (CG) with placebo or CG with bevacizumab at a dose of 7.5 or 15 mg/kg IV infusion day 1 of every 3-week cycle. In the bevacizumab-containing arms, patients could receive bevacizumab as a single-agent every 3 weeks until disease progression or unacceptable toxicity. Trial results show that 94% (277/296) of eligible patients went on to receive single agent bevacizumab at cycle 7. A high proportion of patients (approximately 62%) went on to receive a variety of non-protocol specified anti-cancer therapies, which may have impacted the analysis of overall survival.
The efficacy results are presented in Table 10. (See Table 10.)
Click on icon to see table/diagram/image
First-line treatment of non-squamous NSCLC with EGFR activating mutations in combination with erlotinib: JO25567: Study JO25567 was a randomized, open-label, multi-center Phase II study conducted in Japan to evaluate the efficacy and safety of bevacizumab used in addition to erlotinib in patients with non-squamous NSCLC with EGFR activating mutations (exon 19 deletion or exon 21 L858R mutation) who had not received prior systemic therapy for Stage IIIB/IV or recurrent disease.
The primary endpoint was progression-free survival (PFS) based on independent review assessment. Secondary endpoints included overall survival, response rate, disease control rate, duration of response, and safety.
EGFR mutation status was determined for each patient prior to patient screening and 154 patients were randomised to receive either erlotinib + bevacizumab (erlotinib 150 mg oral daily + bevacizumab [15 mg/kg IV every 3 weeks]) or erlotinib monotherapy (150 mg oral daily) until disease progression (PD) or unacceptable toxicity. In the absence of PD, discontinuation of one component of study treatment in the erlotinib + bevacizumab arm did not lead to discontinuation of the other component of study treatment as specified in the study protocol.
The efficacy results of the study are presented in Table 11. (See Table 11.)
Click on icon to see table/diagram/image
Advanced and/or metastatic renal cell cancer (mRCC): Bevacizumab in combination with interferon alfa-2a for the first-line treatment of advanced and/or metastatic renal cell cancer (BO17705): This was a phase III randomised double-blind trial conducted to evaluate the efficacy and safety of bevacizumab in combination with interferon (IFN) alfa-2a versus IFN alfa-2a alone as first-line treatment in mRCC. The 649 randomised patients (641 treated) had Karnofsky Performance Status (KPS) of ≥ 70%, no central nervous system (CNS) metastases and adequate organ function. Patients were nephrectomised for primary renal cell carcinoma. Bevacizumab 10 mg/kg was given every 2 weeks until disease progression. IFN alfa-2a was given up to 52 weeks or until disease progression at a recommended starting dose of 9 million international units (MIU) three times a week, allowing a dose reduction to 3 MIU three times a week in 2 steps. Patients were stratified according to country and Motzer score and the treatment arms were shown to be well balanced for the prognostic factors.
The primary endpoint was overall survival, with secondary endpoints for the trial included progression-free survival. The addition of bevacizumab to IFN-alpha-2a significantly increased PFS and objective tumour response rate. These results have been confirmed through an independent radiological review. However, the increase in the primary endpoint of overall survival by 2 months was not significant (HR= 0.91). A high proportion of patients (approximately 63% IFN/placebo; 55% bevacizumab/IFN) received a variety of non-specified post-trial anti-cancer therapies, including antineoplastic agents, which may have impacted the analysis of overall survival.
The efficacy results are presented in Table 12. (See Table 12.)
Click on icon to see table/diagram/image
An exploratory multivariate Cox regression model using backward selection indicated that the following baseline prognostic factors were strongly associated with survival independent of treatment: gender, white blood cell count, platelets, body weight loss in the 6 months prior to trial entry, number of metastatic sites, sum of longest diameter of target lesions, Motzer score. Adjustment for these baseline factors resulted in a treatment hazard ratio of 0.78 (95% CI [0.63; 0.96], p=0.0219), indicating a 22% reduction in the risk of death for patients in the bevacizumab + IFN alfa-2a arm compared to IFN alfa-2a arm.
Ninety seven (97) patients in the IFN alfa-2a arm and 131 patients in the bevacizumab arm reduced the dose of IFN alfa-2a from 9 MIU to either 6 or 3 MIU three times a week as pre-specified in the protocol. Dose-reduction of IFN alfa-2a did not appear to affect the efficacy of the combination of bevacizumab and IFN alfa-2a based on PFS event free rates over time, as shown by a sub-group analysis. The 131 patients in the bevacizumab + IFN alfa-2a arm who reduced and maintained the IFN alfa-2a dose at 6 or 3 MIU during the trial, exhibited at 6, 12 and 18 months PFS event free rates of 73, 52 and 21% respectively, as compared to 61, 43 and 17% in the total population of patients receiving bevacizumab + IFN alfa-2a.
AVF2938: This was a randomised, double-blind, phase II clinical trial investigating bevacizumab 10 mg/kg in a 2 weekly schedule with the same dose of bevacizumab in combination with 150 mg daily erlotinib, in patients with metastatic clear cell RCC. A total of 104 patients were randomised to treatment in this trial, 53 to bevacizumab 10 mg/kg every 2 weeks plus placebo and 51 to bevacizumab 10 mg/kg every 2 weeks plus erlotinib 150 mg daily. The analysis of the primary endpoint showed no difference between the bevacizumab + Placebo arm and the bevacizumab + Erlotinib arm (median PFS 8.5 versus 9.9 months). Seven patients in each arm had an objective response. The addition of erlotinib to bevacizumab did not result in an improvement in OS (HR = 1.764; p=0.1789), duration of objective response (6.7 vs 9.1 months) or time to symptom progression (HR = 1.172; p=0.5076).
AVF0890: This was a randomised phase II trial conducted to compare the efficacy and safety of bevacizumab versus placebo. A total of 116 patients were randomised to receive bevacizumab 3 mg/kg every 2 weeks (n=39), 10 mg/kg every 2 weeks; (n=37), or placebo (n=40). An interim analysis showed there was a significant prolongation of time to disease progression in the 10 mg/kg group as compared with the placebo group (hazard ratio, 2.55; p < 0.001). There was a small difference, of borderline significance, between the time to disease progression in the 3 mg/kg group and that in the placebo group (hazard ratio, 1.26; p=0.053). Four patients had objective (partial) response, and all of these had received the 10 mg/kg dose bevacizumab; the ORR for the 10 mg/kg dose was 10%.
Epithelial ovarian, fallopian tube and primary peritoneal cancer: Front-line treatment of ovarian cancer: The safety and efficacy of bevacizumab in the front-line treatment of patients with epithelial ovarian, fallopian tube or primary peritoneal cancer were studied in two phase III trials (GOG-0218 and BO17707) that evaluated the effect of bevacizumab addition to carboplatin and paclitaxel based chemotherapy compared to the chemotherapy alone.
GOG-0218: The GOG-0218 study was a phase III multicentre, randomised, double-blind, placebo-controlled, three arm study evaluating the effect of adding bevacizumab to an approved chemotherapy regimen (carboplatin and paclitaxel) in patients with advanced (FIGO stages IIIB, IIIC and IV) epithelial ovarian, fallopian tube or primary peritoneal cancer.
Patients who had received prior therapy with bevacizumab or prior systemic anticancer therapy for ovarian cancer (e.g. chemotherapy, monoclonal antibody therapy, tyrosine kinase inhibitor therapy, or hormonal therapy) or previous radiotherapy to the abdomen or pelvis were excluded from the study.
A total of 1873 patients were randomised in equal proportions to the following three arms: Carboplatin + paclitaxel + placebo (CPP) arm: Five cycles of placebo (started cycle 2) in combination with carboplatin (AUC 6) and paclitaxel (175 mg/m
2) for 6 cycles followed by placebo alone, for a total of up to 15 months of therapy.
Carboplatin + paclitaxel + bevacizumab (CPB15) arm: Five cycles of bevacizumab (15 mg/kg q3w started cycle 2) in combination with carboplatin (AUC 6) and paclitaxel (175 mg/m
2) for 6 cycles followed by placebo alone, for a total of up to 15 months of therapy.
Carboplatin + paclitaxel + bevacizumab (CPB15+) arm: Five cycles of bevacizumab (15 mg/kg q3w started cycle 2) in combination with carboplatin (AUC 6) and paclitaxel (175 mg/m
2) for 6 cycles followed by continued use of bevacizumab (15 mg/kg q3w) as single agent for a total of up to 15 months of therapy.
The majority of patients included in the study were White (87% in all three arms); the median age was 60 years in CPP and CPB15 arms and 59 years in CPB15+ arm; and 29% of patients in CPP or CPB15 and 26% in CPB15+ were over 65 years of age. Overall approximately 50% of patients had a GOG PS of 0 at baseline, 43% a GOG PS score of 1, and 7% a GOG PS score of 2. Most patients had EOC (82% in CPP and CPB15, 85% in CPB15+) followed by PPC (16% in CPP, 15% in CPB15, 13% in CPB15+) and FTC (fallopian tube cancer) (1% in CPP, 3% in CPB15, 2% in CPB15+). The majority of patients had serious adenocarcinoma histologic type (85% in CPP and CPB15, 86% in CPB15+). Overall approximately 34% of patients were FIGO Stage III optimally debulked with gross residual disease, 40% Stage III sub-optimally debulked, and 26% were Stage IV patients.
The primary endpoint was PFS based on investigator's assessment of disease progression based on radiological scans or CA-125 levels, or symptomatic deterioration per protocol. In addition, a prespecified analysis of the data censoring for CA-125 progression events was conducted, as well as an independent review of PFS as determined by radiological scans.
The trial met its primary objective of PFS improvement. Compared with patients who underwent chemotherapy (carboplatin and paclitaxel) alone in the front-line setting, patients who received bevacizumab at a dose of 15 mg/kg q3w in combination with chemotherapy and continued to receive bevacizumab alone (CPB15+), had a clinically meaningful and statistically significant improvement in PFS.
In patients who only received bevacizumab in combination with chemotherapy and did not continue to receive bevacizumab alone (CPB15), no clinically meaningful benefit in PFS was observed.
The results of this study are summarised in Table 13. (See Table 13.)
Click on icon to see table/diagram/image
Prespecified PFS analyses were conducted, all with a cut-off date of 29 September 2009. The results of these prespecified analyses are as follows: The protocol specified analysis of investigator-assessed PFS (without censoring for CA-125 progression or non-protocol therapy [NPT]) shows a stratified hazard ratio of 0.71 (95% CI: 0.61-0.83, 1-sided log-rank p-value < 0.0001) when CPB15+ is compared with CPP, with a median PFS of 10.4 months in the CPP arm and 14.1 months in the CPB15+ arm.
The primary analysis of investigator-assessed PFS (censoring for CA-125 progressions and NPT) shows a stratified hazard ratio of 0.62 (95% CI: 0.52-0.75, 1-sided log-rank p-value < 0.0001) when CPB15+ is compared with CPP, with a median PFS of 12.0 months in the CPP arm and 18.2 months in the CPB15+ arm.
The analysis of PFS as determined by the independent review committee (censoring for NPT) shows a stratified hazard ratio of 0.62 (95% CI: 0.50-0.77, 1-sided log-rank p-value < 0.0001) when CPB15+ is compared with CPP, with a median PFS of 13.1 in the CPP arm and 19.1 months in the CPB15+ arm.
PFS subgroup analyses by disease stage and debulking status are summarised in Table 14. These results demonstrate robustness of the analysis of PFS as shown in Table 13. (See Table 14.)
Click on icon to see table/diagram/image
BO17707 was a Phase III, two arm, multicentre, randomised, controlled, open-label study comparing the effect of adding bevacizumab to carboplatin plus paclitaxel in patients with FIGO stage I or IIA (Grade 3 or clear cell histology only; n = 142), or FIGO stage IIB - IV (all Grades and all histological types, n = 1386) epithelial ovarian, fallopian tube or primary peritoneal cancer following surgery (NCI-CTCAE v.3).
Patients who had received prior therapy with bevacizumab or prior systemic anticancer therapy for ovarian cancer (e.g. chemotherapy, monoclonal antibody therapy, tyrosine kinase inhibitor therapy, or hormonal therapy) or previous radiotherapy to the abdomen or pelvis were excluded from the study.
A total of 1528 patients were randomised in equal proportions to the following two arms: CP arm: Carboplatin (AUC 6) and paclitaxel (175 mg/m
2) for 6 cycles of 3 weeks duration; CPB7.5+ arm: Carboplatin (AUC 6) and paclitaxel (175 mg/m
2) for 6 cycles of 3 weeks plus bevacizumab (7.5 mg/kg q3w) for up to 12 months (bevacizumab was started at cycle 2 of chemotherapy if treatment was initiated within 4 weeks of surgery or at cycle 1 if treatment was initiated more than 4 weeks after surgery).
The majority of patients included in the study were White (96%), the median age was 57 years in both treatment arms, 25% of patients in each treatment arm were 65 years of age or over, and approximately 50% of patients had an ECOG PS of 1; 7% of patients in each treatment arm had an ECOG PS of 2. The majority of patients had EOC (epithelial ovarian cancer) (87.7%) followed by PPC (primary peritoneal cancer) (6.9%) and FTC (fallopian tube cancer) (3.7%) or a mixture of the three origins (1.7%). Most patients were FIGO Stage III (both 68%) followed by FIGO Stage IV (13% and 14%), FIGO Stage II (10% and 11%) and FIGO Stage I (9% and 7%). The majority of the patients in each treatment arm (74% and 71%) had poorly differentiated (Grade 3) primary tumours at baseline. The incidence of each histologic sub-type of EOC was similar between the treatment arms; 69% of patients in each treatment arm had serious adenocarcinoma histologic type.
The primary endpoint was PFS as assessed by the investigator using RECIST (Response Evaluation Criteria in Solid Tumors).
The trial met its primary objective of PFS improvement. Compared to patients treated with chemotherapy (carboplatin and paclitaxel) alone in the front-line setting, patients who received bevacizumab at a dose of 7.5 mg/kg q3w in combination with chemotherapy and continued to receive bevacizumab for up to 18 cycles had a statistically significant improvement in PFS.
The results of this study are summarised in Table 15. (See Table 15.)
Click on icon to see table/diagram/image
The primary analysis of investigator-assessed PFS with a data cut-off date of 28 February 2010 shows an unstratified hazard ratio of 0.79 (95% CI: 0.68-0.91, 2-sided log-rank p-value 0.0010) with a median PFS of 16.0 months in the CP arm and 18.3 months in the CPB7.5+ arm.
PFS subgroup analyses by disease stage and debulking status are summarized in Table 16. These results demonstrate robustness of the primary analysis of PFS as shown in Table 15. (See Table 16.)
Click on icon to see table/diagram/image
Recurrent ovarian cancer: The safety and efficacy of bevacizumab in the treatment of recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer was studied in two phase III trials (AVF4095g and MO22224) with different patient populations and chemotherapy regimens.
AVF4095g evaluated the efficacy and safety of bevacizumab in combination with carboplatin and gemcitabine in patients with platinum-sensitive recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer.
MO22224 evaluated the efficacy and safety of bevacizumab in combination with paclitaxel, topotecan, or pegylated liposomal doxorubicin in patients with platinum-resistant recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer.
AVF4095g: The safety and efficacy of bevacizumab in the treatment of patients with platinum-sensitive, recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer, who have not received prior chemotherapy in the recurrent setting or prior bevacizumab treatment, was studied in a phase III randomised, double-blind, placebo-controlled trial (AVF4095g). The study compared the effect of adding bevacizumab to carboplatin and gemcitabine chemotherapy and continuing bevacizumab as a single agent until disease progression, to carboplatin and gemcitabine alone.
Only patients with histologically documented ovarian, primary peritoneal, or fallopian tube carcinoma that had recurred > 6 months after platinum-based chemotherapy and who had not received chemotherapy in the recurrent setting and who had not received prior therapy with bevacizumab or other VEGF inhibitors or VEGF receptor-targeted agents were included in the study.
A total of 484 patients with measurable disease were randomised 1:1 to either: Carboplatin (AUC4, Day 1) and gemcitabine (1000 mg/m
2 on Days 1 and 8) and concurrent placebo every 3 weeks for 6 and up to 10 cycles followed by placebo (every 3 weeks) alone until disease progression or unacceptable toxicity; Carboplatin (AUC4, Day 1) and gemcitabine (1000 mg/m
2 on Days 1 and 8) and concurrent bevacizumab (15 mg/kg Day 1) every 3 weeks for 6 and up to 10 cycles followed by bevacizumab (15 mg/kg every 3 weeks) alone until disease progression or unacceptable toxicity.
The primary endpoint was PFS based on investigator assessment using modified RECIST 1.0. Additional endpoints included objective response, duration of response, overall survival and safety. An independent review of the primary endpoint was also conducted.
The results of this study are summarized in Table 17. (See Table 17.)
Click on icon to see table/diagram/image
PFS subgroup analyses depending on recurrence since last platinum therapy are summarised in Table 18. (See Table 18.)
Click on icon to see table/diagram/image
MO22224: Study MO22224 evaluated the efficacy and safety of bevacizumab in combination with chemotherapy for platinum-resistant recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer. This study was designed as an open-label, randomized, two-arm Phase III evaluation of bevacizumab plus chemotherapy (CT+BV) versus chemotherapy alone (CT).
A total of 361 patients were enrolled into this study and administered either chemotherapy (paclitaxel, topotecan, or pegylated liposomal doxorubicin (PLD) alone or in combination with bevacizumab: CT Arm (chemotherapy alone): Paclitaxel 80 mg/m
2 as a 1-hour IV infusion on Days 1, 8, 15, and 22 every 4 weeks; Topotecan 4 mg/m
2 as a 30-minute IV infusion on Days 1, 8, and 15 every 4 weeks. Alternatively, a 1.25 mg/m
2 dose could be administered over 30 minutes on Days 1-5 every 3 weeks; PLD 40 mg/m
2 as a 1 mg/min IV infusion on Day 1 only every 4 weeks. After Cycle 1, the drug could be delivered as a 1-hour infusion.
CT+BV Arm (chemotherapy plus bevacizumab): The chosen chemotherapy was combined with bevacizumab 10 mg/kg IV infusion every 2 weeks (or bevacizumab 15 mg/kg every 3 weeks if used in combination with topotecan 1.25 mg/m
2 on Days 1-5 every 3 weeks).
Eligible patients had epithelial ovarian, fallopian tube or primary peritoneal cancer that progressed within <6 months of previous platinum therapy consisting of a minimum of 4 platinum therapy cycles. Patients should have had a life expectancy of ≥ 12 weeks and no prior radiotherapy to the pelvis or abdomen. Most patients were FIGO Stage IIIC or Stage IV. The majority of patients in both arms had an ECOG PS of 0 (CT: 56.4% vs. CT + BV: 61.2%). The percentage of patients with an ECOG PS of 1 or ≥ 2 was 38.7% and 5.0% in the CT arm, and 29.8% and 9.0% in the CT + BV arm. Information on race exists for 29.3% of patients and nearly all patients were white. The median age of patients was 61.0 (range: 25-84) years. A total of 16 patients (4.4%) were > 75 years old. The overall rates of discontinuation due to adverse events were 8.8% in the CT arm and 43.6% in the CT + BV arm (mostly due to Grade 2-3 adverse events) and the median time to discontinuation in the CT + BV arm was 5.2 months compared with 2.4 months in the CT arm. The rates of discontinuation due to adverse events in the subgroup of patients > 65 years old were 8.8% in the CT arm and 50.0% in the CT + BV arm. The HR for PFS was 0.47 (95% CI: 0.35, 0.62) and 0.45 (95% CI: 0.31, 0.67) for the < 65 and ≥ 65 subgroups, respectively.
The primary endpoint was progression-free-survival, with secondary endpoints including objective response rate and overall survival. Results are presented in Table 19. (See Table 19.)
Click on icon to see table/diagram/image
The trial met its primary objective of PFS improvement. Compared to patients treated with chemotherapy (paclitaxel, topotecan or PLD) alone in the recurrent platinum-resistant setting, patients who received bevacizumab at a dose of 10 mg/kg every 2 weeks (or 15 mg/kg every 3 weeks if used in combination with 1.25 mg/m
2 topotecan on Days 1-5 every 3 weeks) in combination with chemotherapy and continued to receive bevacizumab until disease progression or unacceptable toxicity, had a statistically significant improvement in PFS. The exploratory PFS and OS analyses by chemotherapy cohort (paclitaxel, topotecan and PLD) are summarized in Table 20. (See Table 20.)
Click on icon to see table/diagram/image
Glioblastoma: AVF3708g: The efficacy and safety of bevacizumab as treatment for patients with glioblastoma was studied in an open-label, multicentre, randomised, non-comparative study (AVF3708g).
Glioblastoma patients in first or second relapse after prior radiotherapy (completed at least 8 weeks prior to receiving bevacizumab) and temozolomide, were randomised (1:1) to receive bevacizumab (10 mg/kg IV infusion every 2 weeks) or bevacizumab plus irinotecan (125 mg/m
2 IV or 340 mg/m
2 IV for patients on enzyme-inducing anti-epileptic drugs every 2 weeks) until disease progression or until unacceptable toxicity. The primary endpoints of the study were 6-month PFS and objective response rate as assessed by an independent review facility (IRF). Other outcome measures were duration of PFS, duration of response and overall survival.
Results of the study are summarised in Table 21. (See Table 21.)
Click on icon to see table/diagram/image
In study AVF3708g, six-month PFS based on IRF assessments was significantly higher (p < 0.0001) compared with historical controls for both treatment arms: 42.6% in the bevacizumab arm and 50.3% in the bevacizumab plus irinotecan arm (investigator assessment: 43.6% in the bevacizumab arm and 57.9% in the bevacizumab plus irinotecan arm). Objective response rates were also significantly higher (p < 0.0001) compared with historical controls for both treatment arms: 28.2% in the bevacizumab arm and 37.8% in the bevacizumab plus irinotecan arm (investigator assessment: 41.2% in the bevacizumab arm and 51.2% in the bevacizumab plus irinotecan arm).
The majority of patients who were receiving steroids at baseline, including responders and non-responders, were able to reduce their steroid utilization over time while receiving bevacizumab treatment. The majority of patients experiencing an objective response or prolonged PFS (at week 24) were able to maintain or improve their neurocognitive functions while on study treatment compared to baseline. The majority of patients that remained in the study and were progression free at 24 weeks, had a KPS that remained stable.
Cervical Cancer: GOG-0240: The efficacy and safety of bevacizumab in combination with chemotherapy (paclitaxel and cisplatin or paclitaxel and topotecan) in the treatment for patients with persistent, recurrent or metastatic carcinoma of the cervix was evaluated in study GOG-0240, a randomised, four-arm, open label, multi-centre phase III trial.
A total of 452 patients were randomised to receive either: Paclitaxel 135 mg/m
2 IV infusion over 24 hours on Day 1 and cisplatin 50 mg/m
2 IV infusion on Day 2, every 3 weeks (q3w); or Paclitaxel 175 mg/m
2 IV infusion over 3 hours on Day 1 and cisplatin 50 mg/m
2 IV infusion on Day 2 (q3w); or Paclitaxel 175 mg/m
2 IV infusion over 3 hours on Day 1 and cisplatin 50 mg/m
2 IV infusion on Day 1 (q3w).
Paclitaxel 135 mg/m
2 IV infusion over 24 hours on Day 1 and cisplatin 50 mg/m
2 IV infusion on Day 2 plus bevacizumab 15 mg/kg IV infusion on Day 2 (q3w); or Paclitaxel 175 mg/m
2 IV infusion over 3 hours on Day 1 and cisplatin 50 mg/m
2 IV infusion on Day 2 plus bevacizumab 15 mg/kg IV infusion on Day 2 (q3w); or Paclitaxel 175 mg/m
2 IV infusion over 3 hours on Day 1 and cisplatin 50 mg/m
2 IV infusion on Day 1 plus bevacizumab 15 mg/kg IV infusion on Day 1 (q3w).
Paclitaxel 175 mg/m
2 IV infusion over 3 hours on Day 1 and topotecan 0.75 mg/m
2 IV infusion over 30 minutes on days 1-3 (q3w).
Paclitaxel 175 mg/m
2 IV infusion over 3 hours on Day 1 and topotecan 0.75 mg/m
2 IV infusion over 30 minutes on Days 1-3 plus bevacizumab 15 mg/kg IV infusion on Day 1 (q3w).
Eligible patients had persistent, recurrent or metastatic squamous cell carcinoma, adenosquamous carcinoma, or adenocarcinoma of the cervix which was not amenable to curative treatment with surgery and/or radiation therapy and who had not received prior therapy with bevacizumab or other VEGF inhibitors or VEGF receptor-targeted agents.
The median age was 46.0 years (range: 20-83) in the chemotherapy alone group and 48.0 years (range: 22-85) in the chemotherapy + bevacizumab group; with 9.3% of patients in the chemotherapy alone group and 7.5% of patients in the chemotherapy + bevacizumab group over the age of 65 years.
Of the 452 patients randomized at baseline, the majority of patients were white (80.0% in the chemotherapy alone group and 75.3% in the chemotherapy + bevacizumab group), had squamous cell carcinoma (67.1% in the chemotherapy alone group and 69.6% in the chemotherapy + bevacizumab group), had persistent/recurrent disease (83.6% in the chemotherapy alone group and 82.8% in the chemotherapy + bevacizumab group), had 1-2 metastatic sites (72.0% in the chemotherapy alone group and 76.2% in the chemotherapy + bevacizumab group), had lymph node involvement (50.2% in the chemotherapy alone group and 56.4% in the chemotherapy + bevacizumab group), and had a platinum free interval ≥ 6 months (72.5% in the chemotherapy alone group and 64.4% in the chemotherapy + bevacizumab group).
The primary efficacy endpoint was overall survival. Secondary efficacy endpoints included PFS and objective response rate. Results from the primary analysis and the follow-up analysis are presented by bevacizumab Treatment and by Trial Treatment in Table 22 and Table 23, respectively. (See Tables 22 and 23.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Paediatric population: The European Medicines Agency has waived the obligation to submit the results of studies, in all subsets of the paediatric population, in breast carcinoma, adenocarcinoma of the colon and rectum, lung carcinoma (small cell and non-small cell carcinoma), kidney and renal pelvis carcinoma (excluding nephroblastoma, nephroblastomatosis, clear cell sarcoma, mesoblastic nephroma, renal medullary carcinoma and rhabdoid tumour of the kidney), ovarian carcinoma (excluding rhabdomyosarcoma and germ cell tumours), fallopian tube carcinoma (excluding rhabdomyosarcoma and germ cell tumours), peritoneal carcinoma (excluding blastomas and sarcomas) and cervix and corpus uteri carcinoma.
Anti-tumour activity was not observed in two studies among a total of 30 children aged > 3 years old with relapsed or progressive high-grade glioma when treated with bevacizumab and irinotecan. There is insufficient information to determine the safety and efficacy of bevacizumab in children with newly-diagnosed high-grade glioma.
In a single-arm study (PBTC-022), 18 children with recurrent or progressive non-pontine high-grade glioma (including 8 with glioblastoma [WHO Grade IV], 9 with anaplastic astrocytoma [Grade III] and 1 with anaplastic oligodendroglioma [Grade III]) were treated with bevacizumab (10 mg/kg) two weeks apart and then with bevacizumab in combination with CPT-11 (125-350 mg/m
2) once every two weeks until progression. There were no objective (partial or complete) radiological responses (MacDonald criteria). Toxicity and adverse reactions included arterial hypertension and fatigue as well as CNS ischaemia with acute neurological deficit.
In a retrospective single institution series, 12 consecutive (2005 to 2008) children with relapsed or progressive high-grade glioma (3 with WHO Grade IV, 9 with Grade III) were treated with bevacizumab (10 mg/kg) and irinotecan (125 mg/m
2) every 2 weeks. There were no complete responses and 2 partial responses (MacDonald criteria).
Pharmacokinetics: The pharmacokinetic data for bevacizumab are available from ten clinical trials in patients with solid tumours. In all clinical trials, bevacizumab was administered as an IV infusion. The rate of infusion was based on tolerability, with an initial infusion duration of 90 minutes. The pharmacokinetics of bevacizumab was linear at doses ranging from 1 to 10 mg/kg.
Distribution: The typical value for central volume (V
c) was 2.73 L and 3.28 L for female and male patients respectively, which is in the range that has been described for IgGs and other monoclonal antibodies. The typical value for peripheral volume (V
p) was 1.69 L and 2.35 L for female and male patients respectively, when bevacizumab is co-administered with anti-neoplastic agents. After correcting for body weight, male patients had a larger V
c (+ 20%) than female patients.
Biotransformation: Assessment of bevacizumab metabolism in rabbits following a single IV dose of
125I-bevacizumab indicated that its metabolic profile was similar to that expected for a native IgG molecule which does not bind VEGF. The metabolism and elimination of bevacizumab is similar to endogenous IgG i.e. primarily via proteolytic catabolism throughout the body, including endothelial cells, and does not rely primarily on elimination through the kidneys and liver. Binding of the IgG to the FcRn receptor results in protection from cellular metabolism and the long terminal half-life.
Elimination: The value for clearance is, on average, equal to 0.188 and 0.220 L/day for female and male patients, respectively. After correcting for body weight, male patients had a higher bevacizumab clearance (+ 17%) than females. According to the two-compartmental model, the elimination half-life is 18 days for a typical female patient and 20 days for a typical male patient.
Low albumin and high tumour burden are generally indicative of disease severity. Bevacizumab clearance was approximately 30% faster in patients with low levels of serum albumin and 7% faster in subjects with higher tumour burden when compared with a typical patient with median values of albumin and tumour burden.
Pharmacokinetics in special populations: The population pharmacokinetics were analysed to evaluate the effects of demographic characteristics. The results showed no significant difference in the pharmacokinetics of bevacizumab in relation to age.
Renal impairment: No trials have been conducted to investigate the pharmacokinetics of bevacizumab in renally impaired patients since the kidneys are not a major organ for bevacizumab metabolism or excretion.
Hepatic impairment: No trials have been conducted to investigate the pharmacokinetics of bevacizumab in patients with hepatic impairment since the liver is not a major organ for bevacizumab metabolism or excretion.
Paediatric population: The pharmacokinetics of bevacizumab have been studied in a limited number of paediatric patients. The resulting pharmacokinetic data suggest that the volume of distribution and clearance of bevacizumab were comparable to that in adults with solid tumours.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image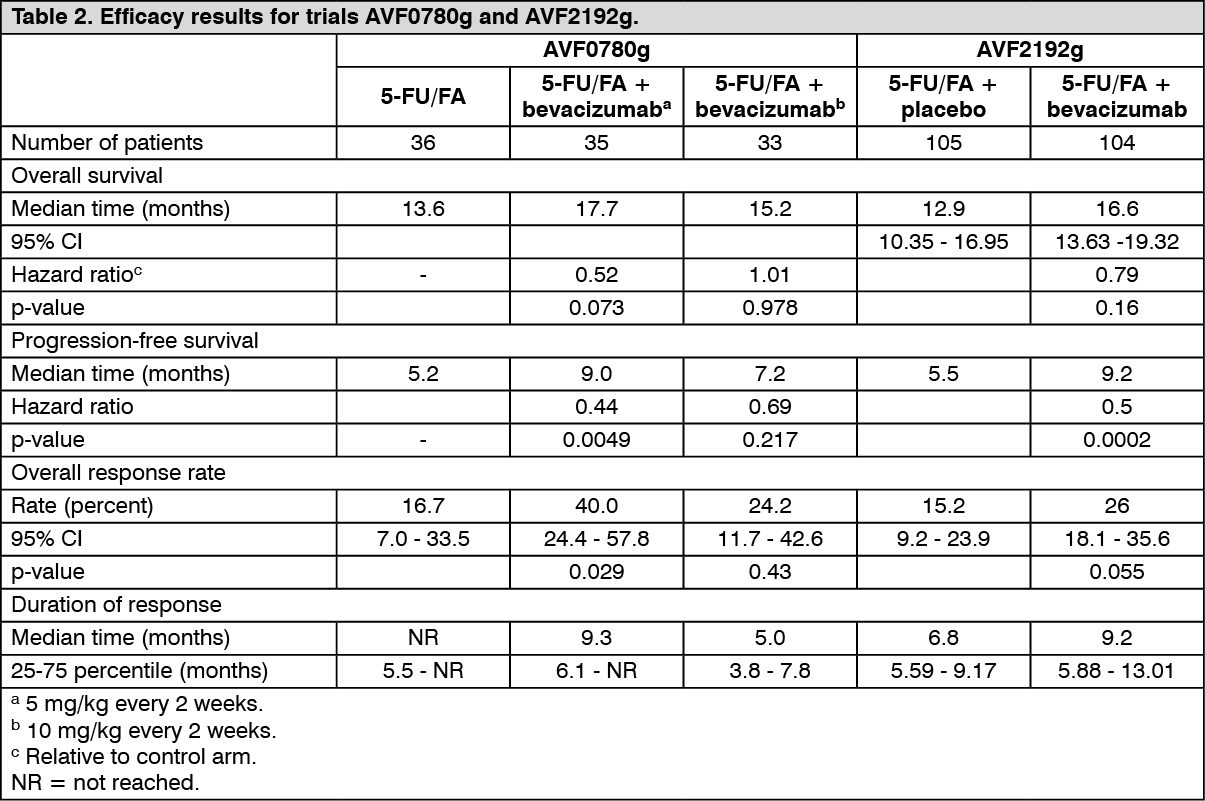 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image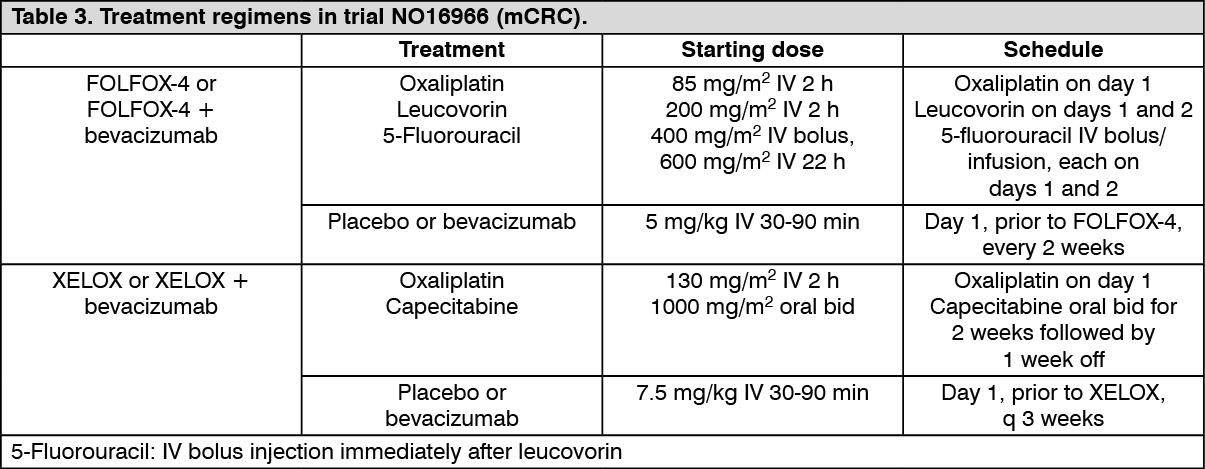 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image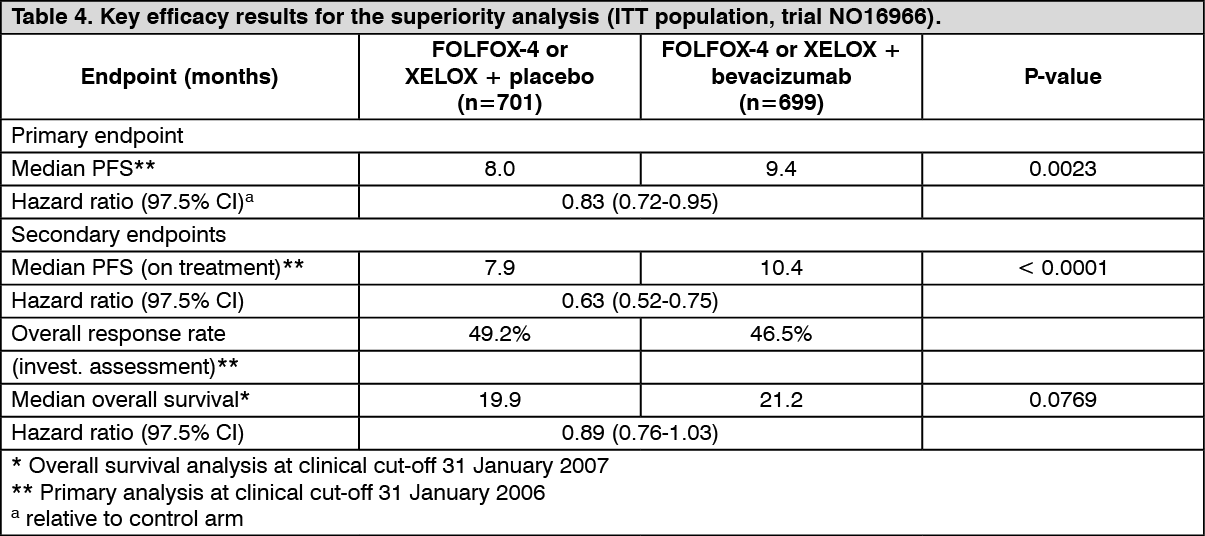 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image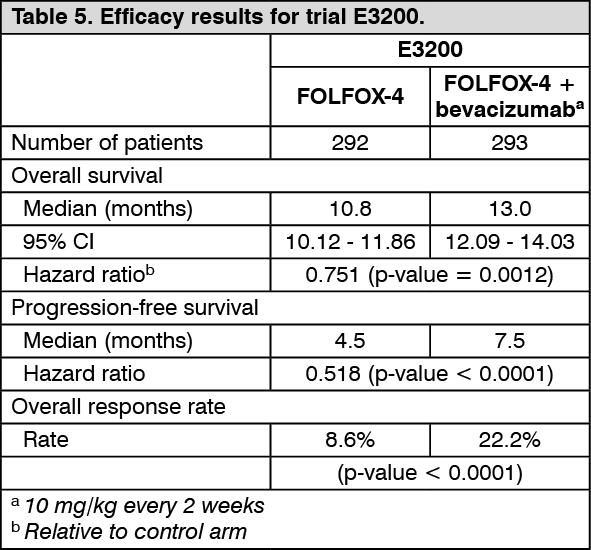 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image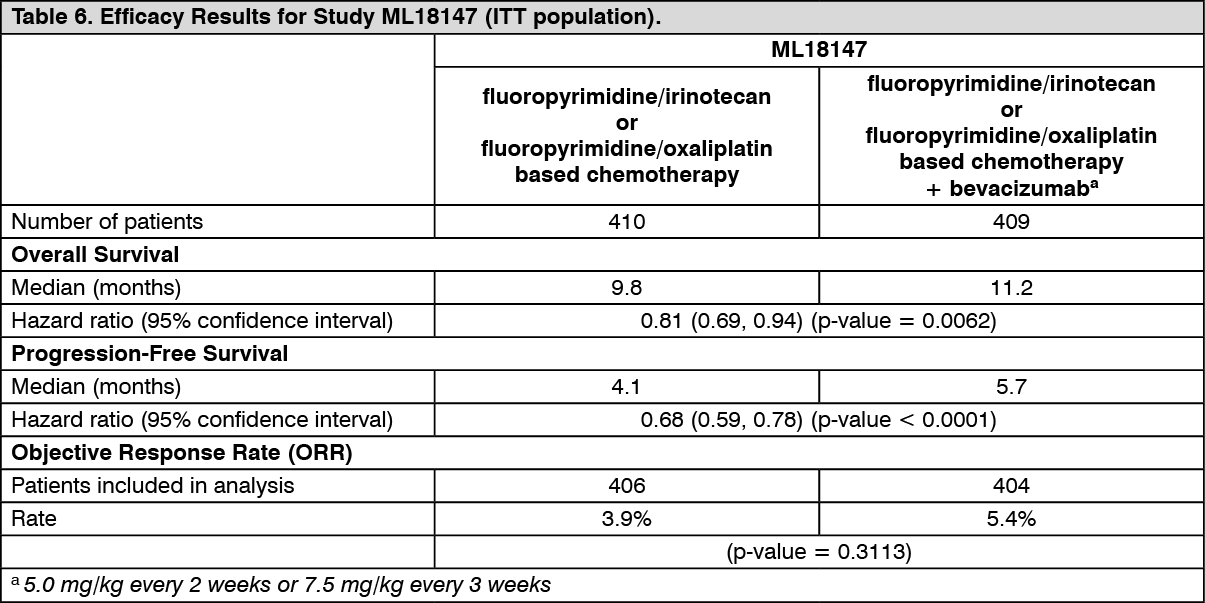 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image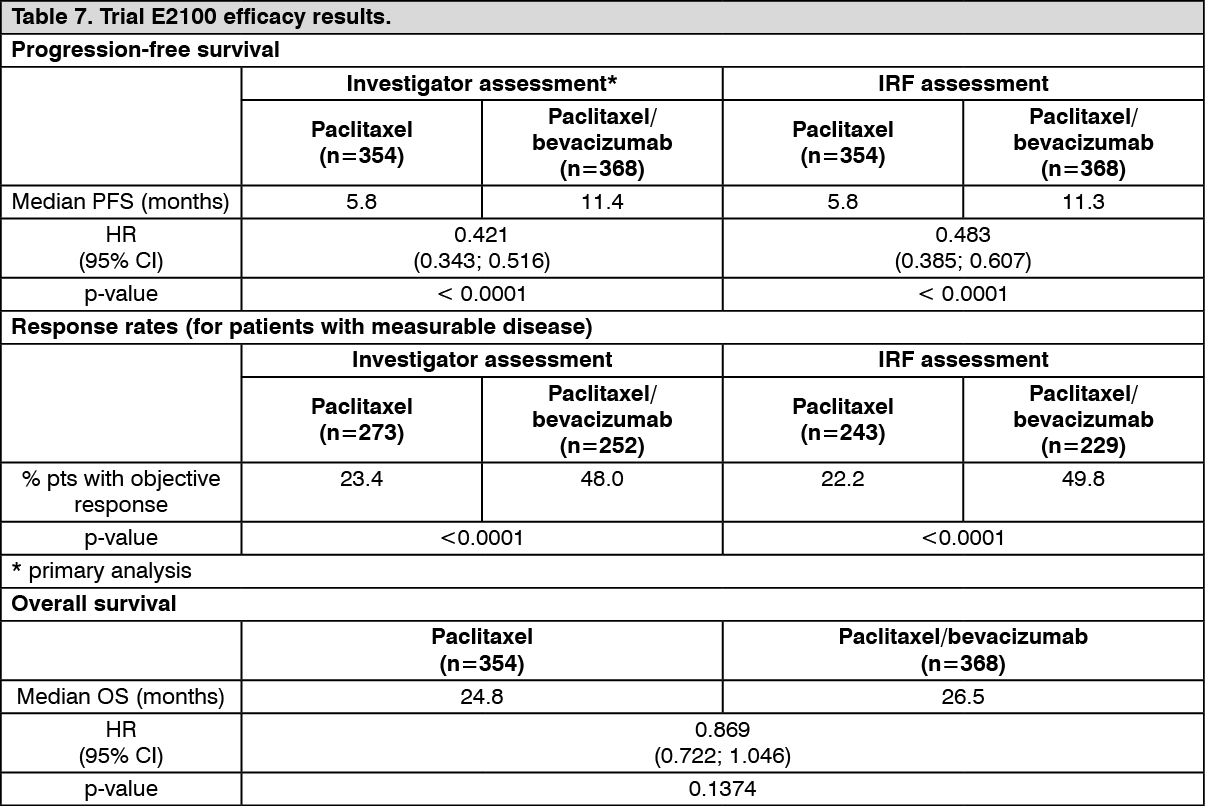 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image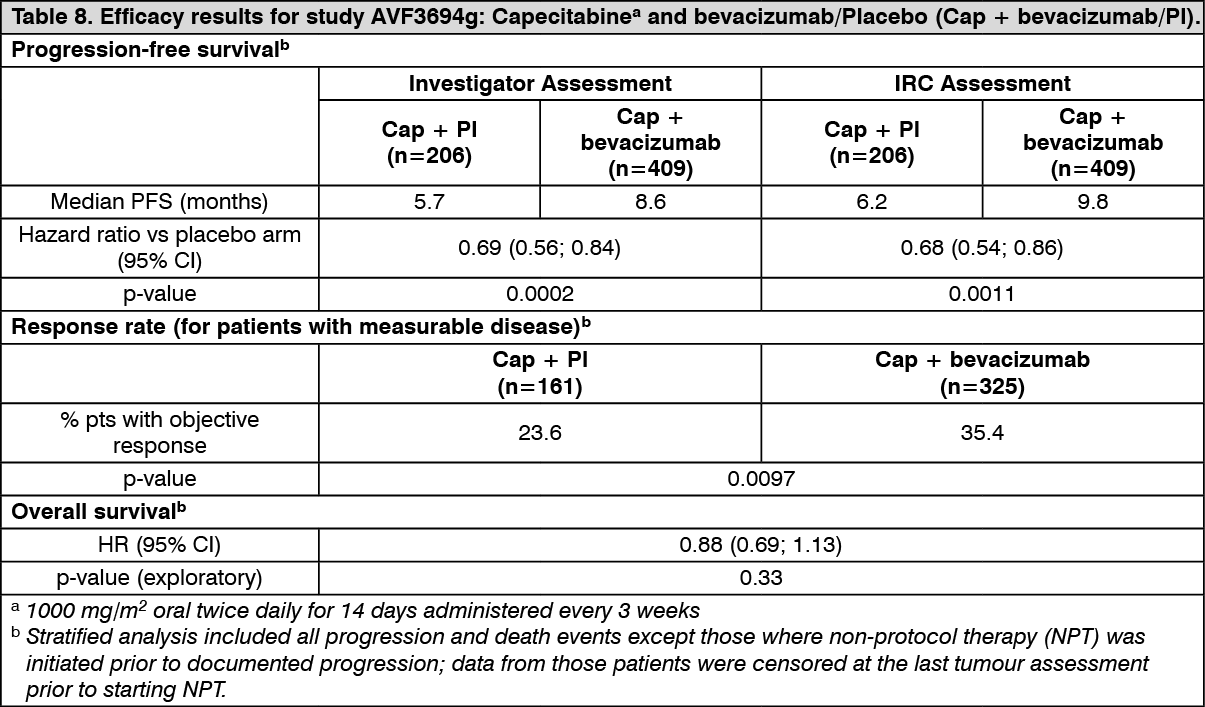 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image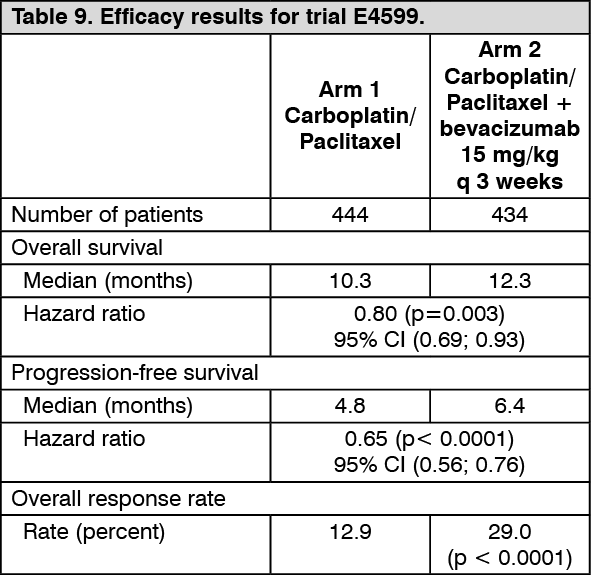 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image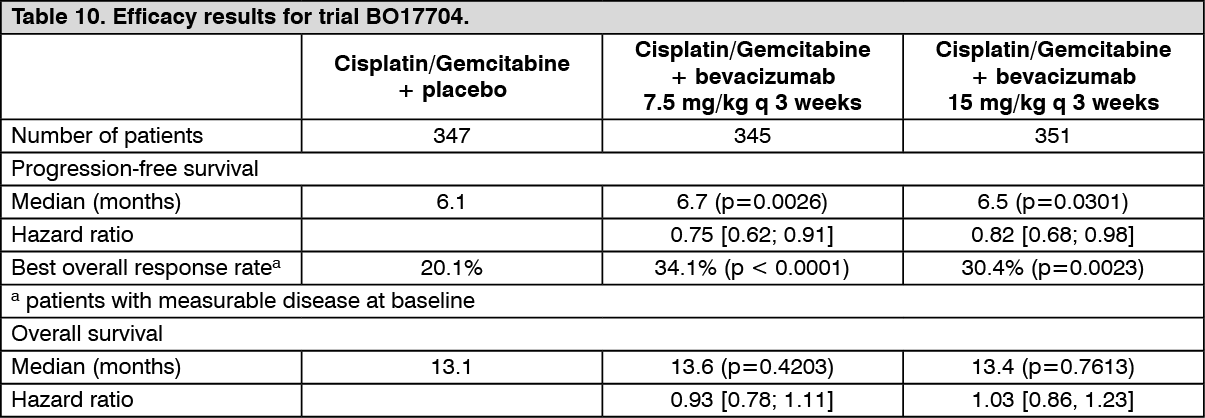 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image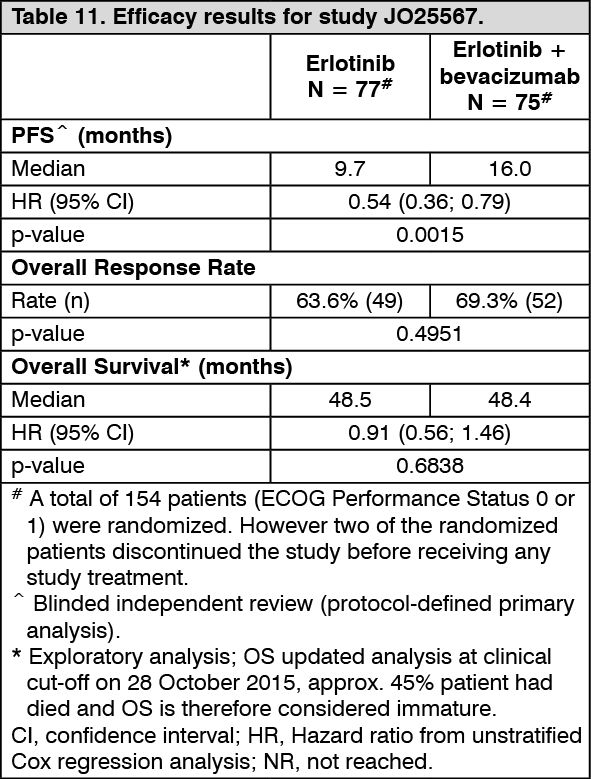 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image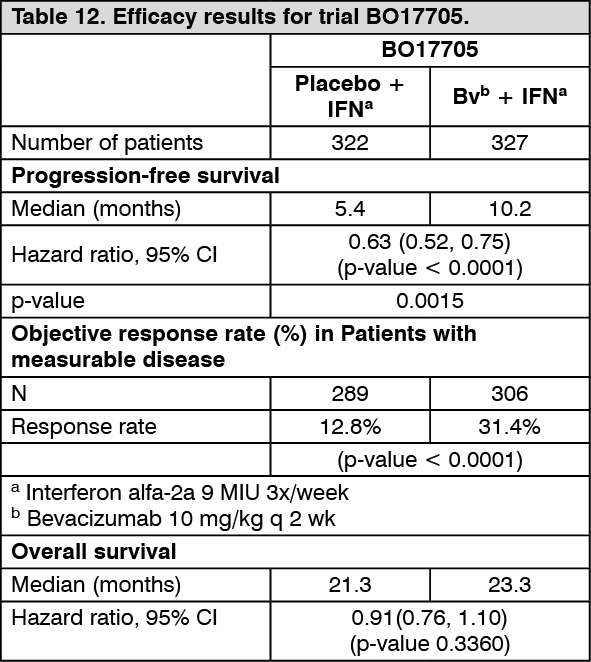 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image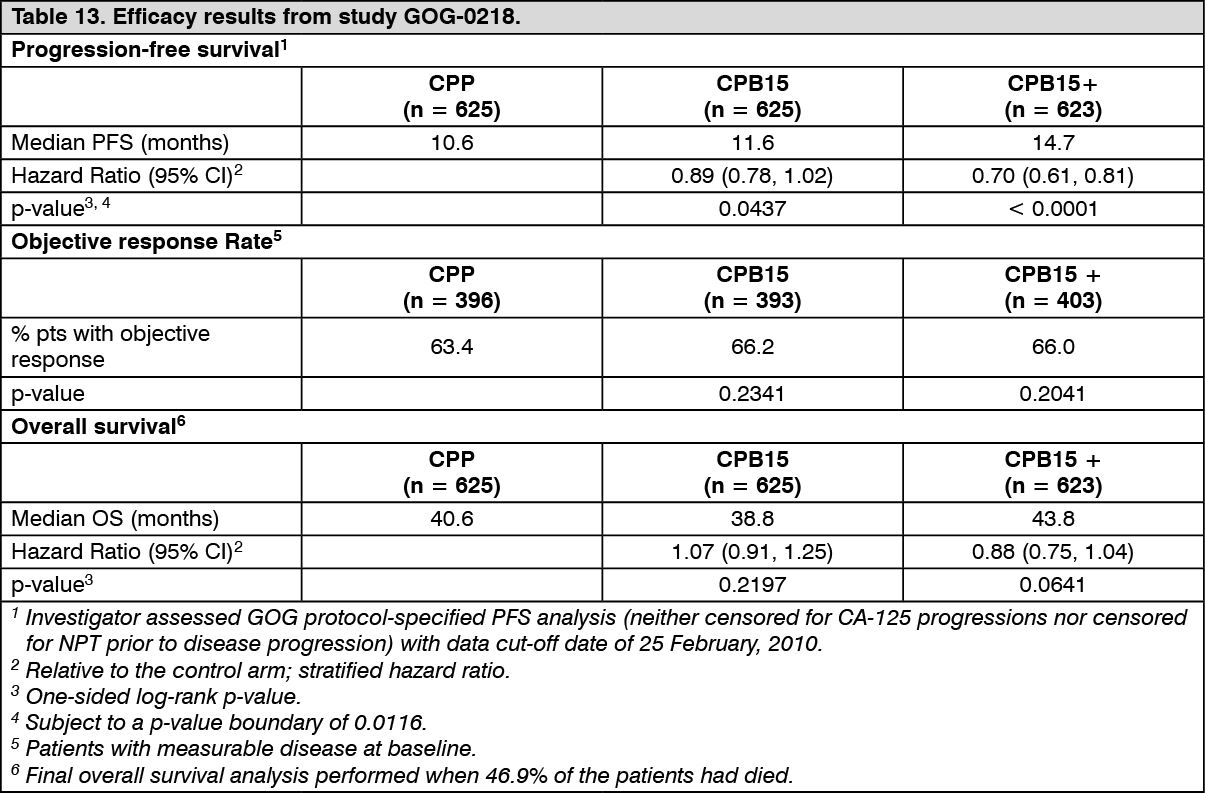 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image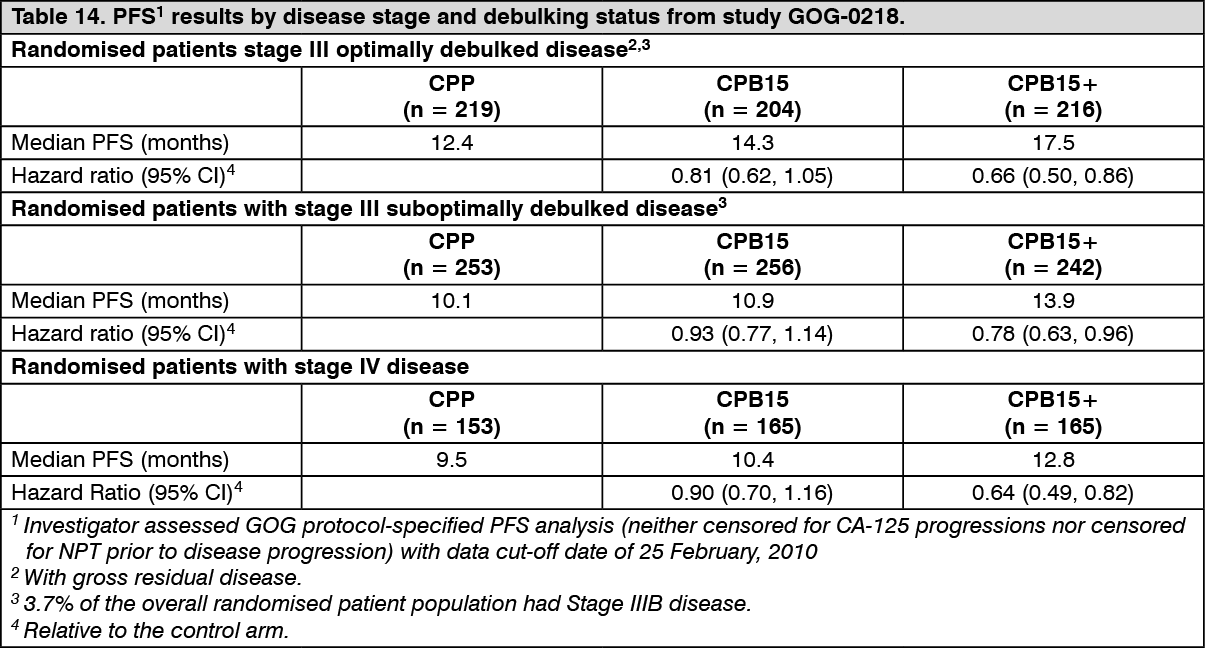 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image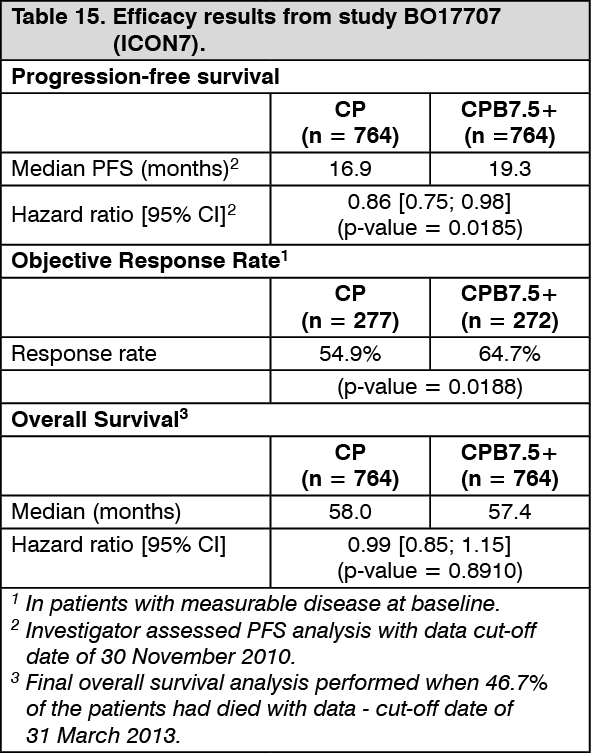 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image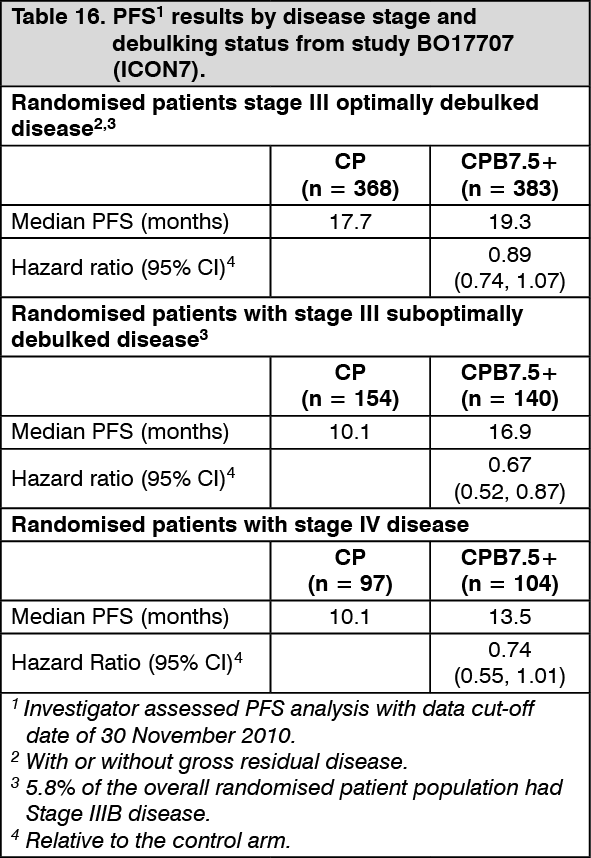 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image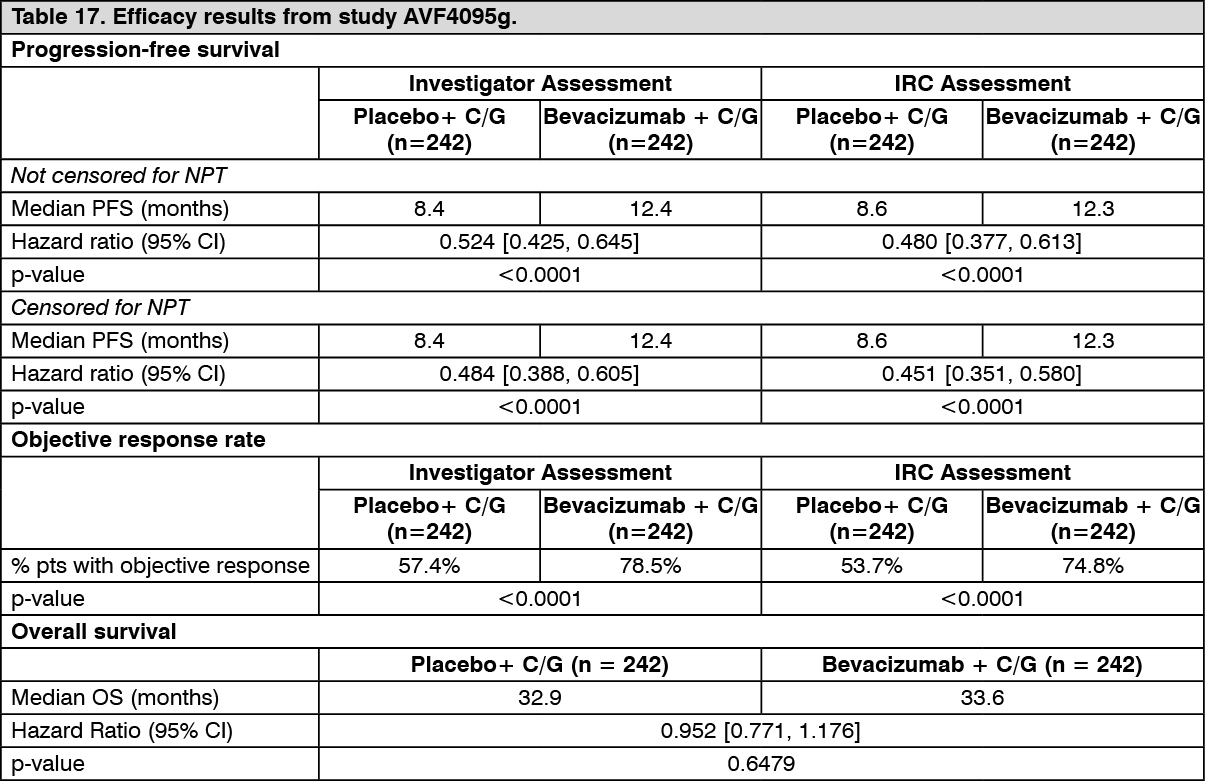 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image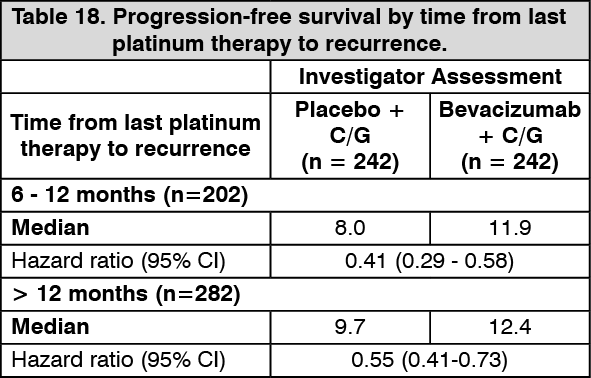 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image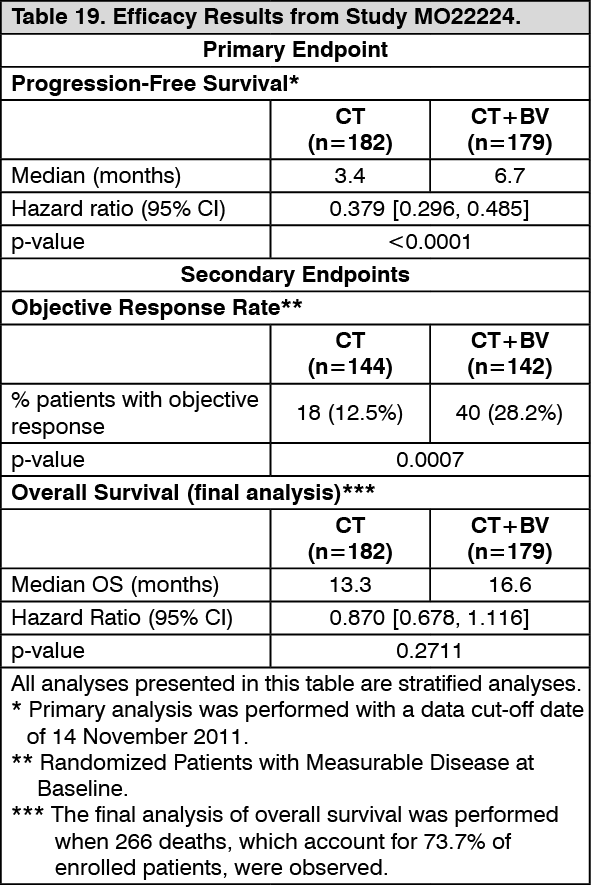 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image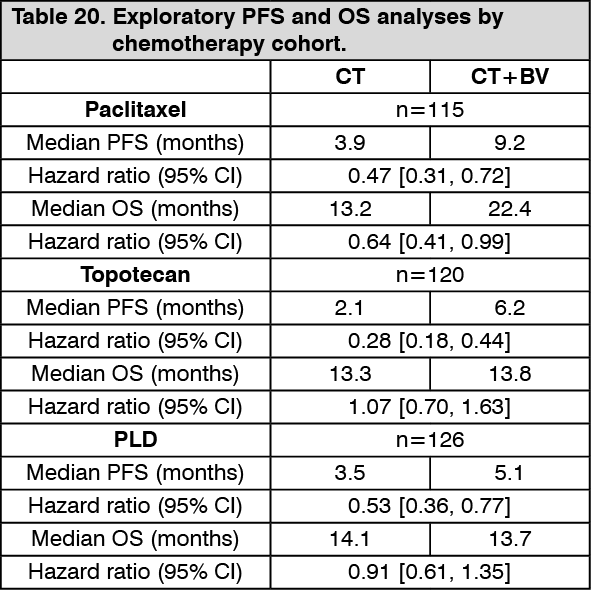 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image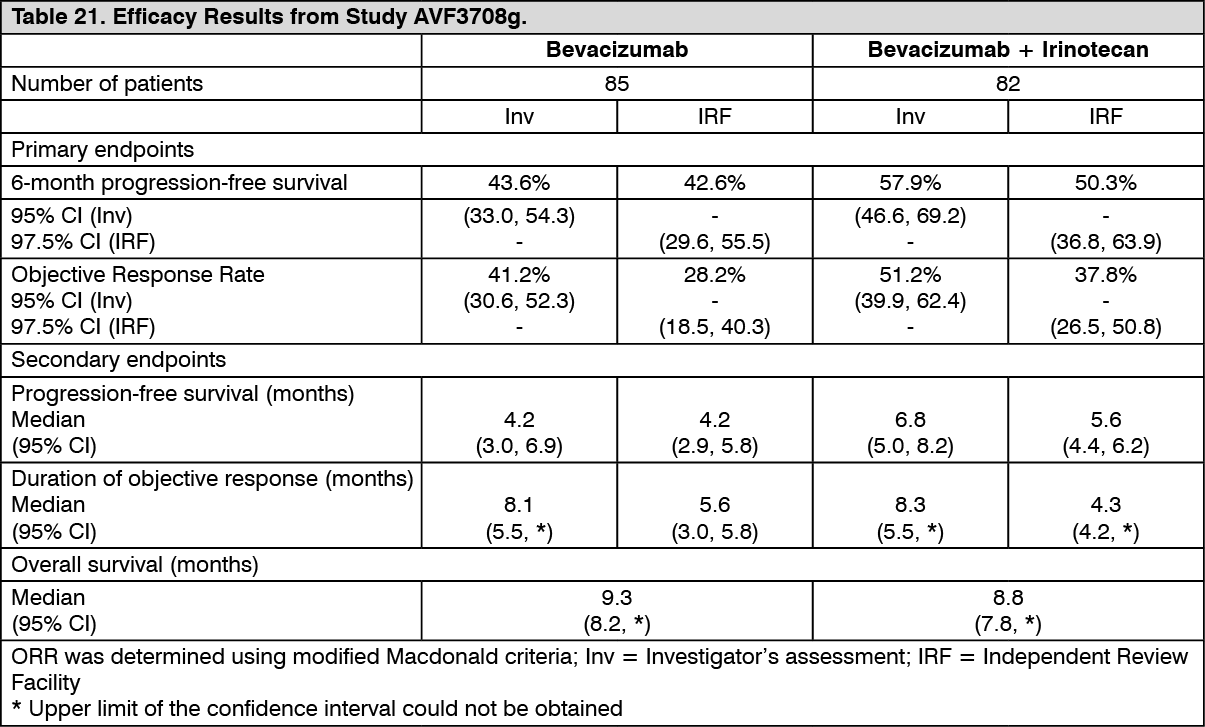 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image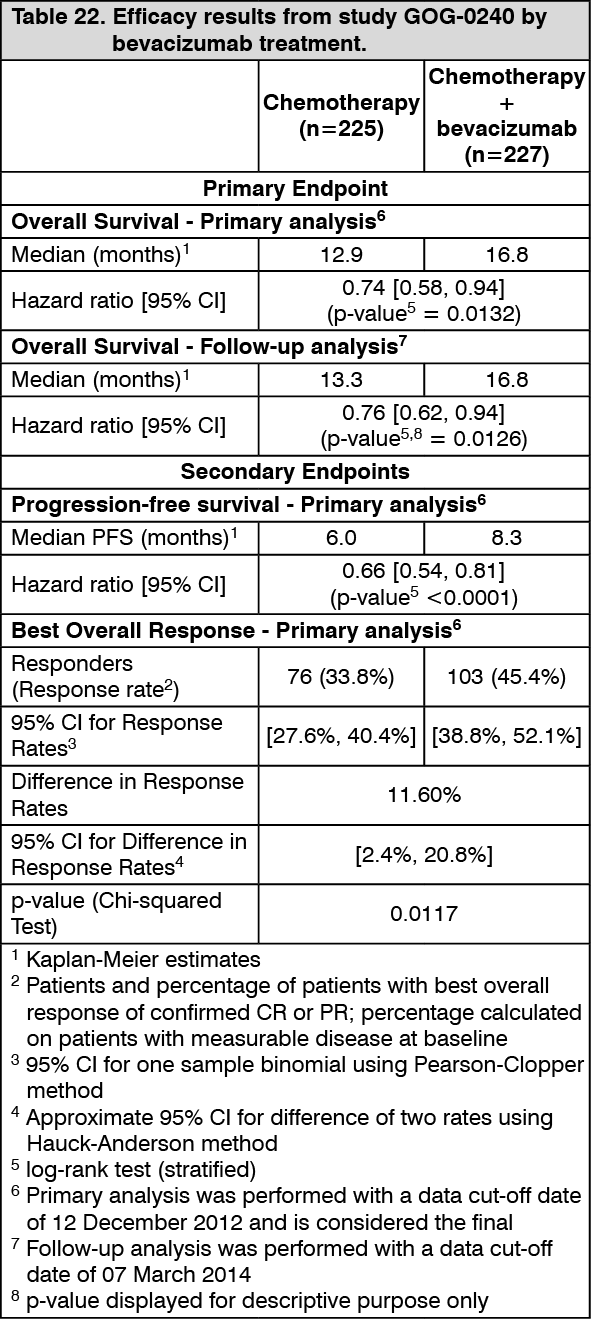 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image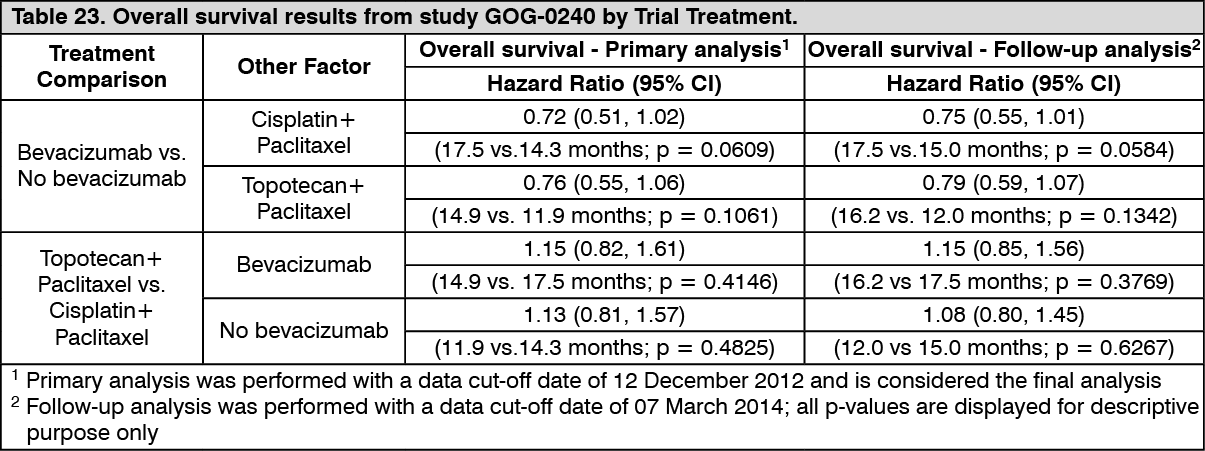 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image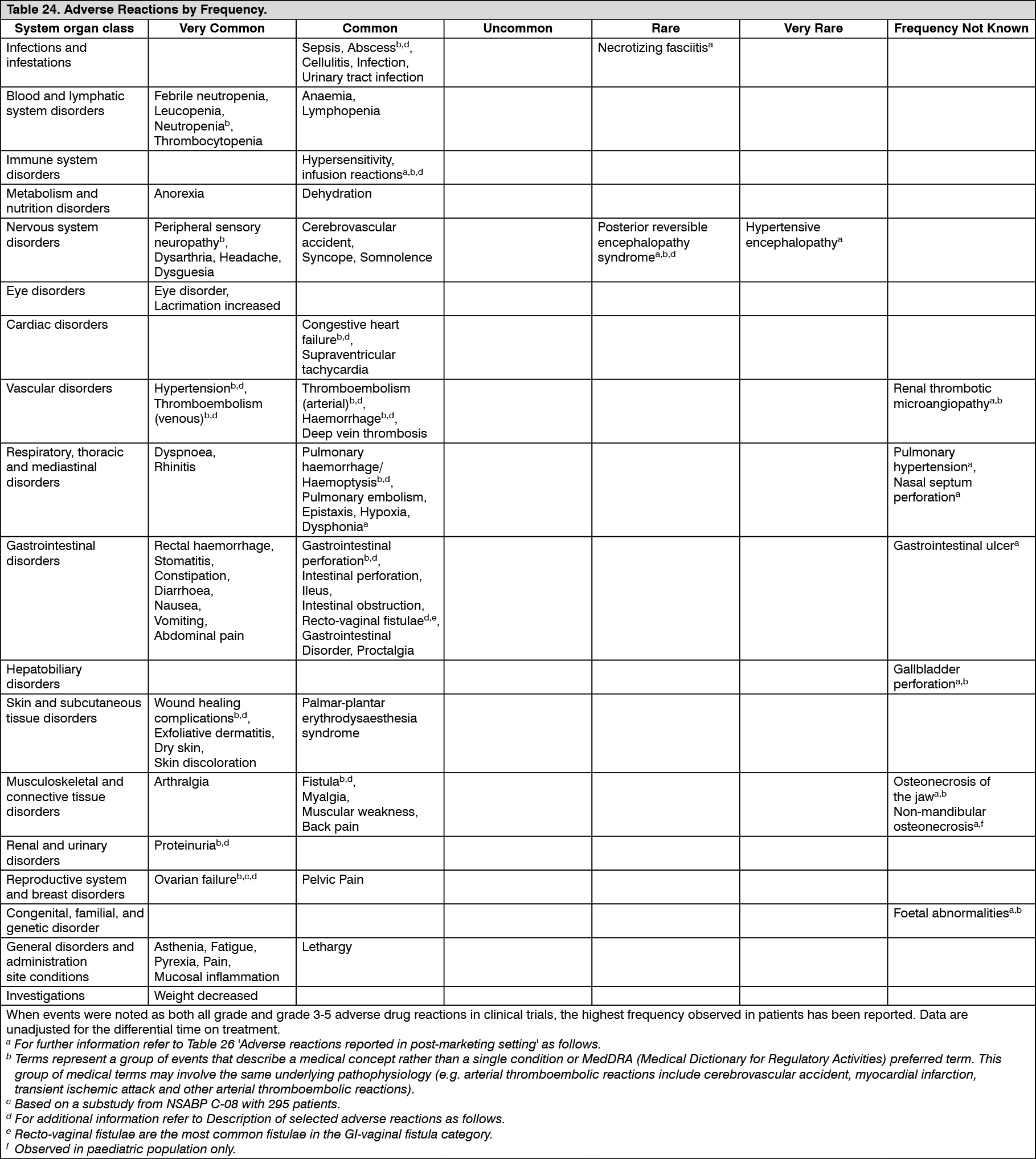 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image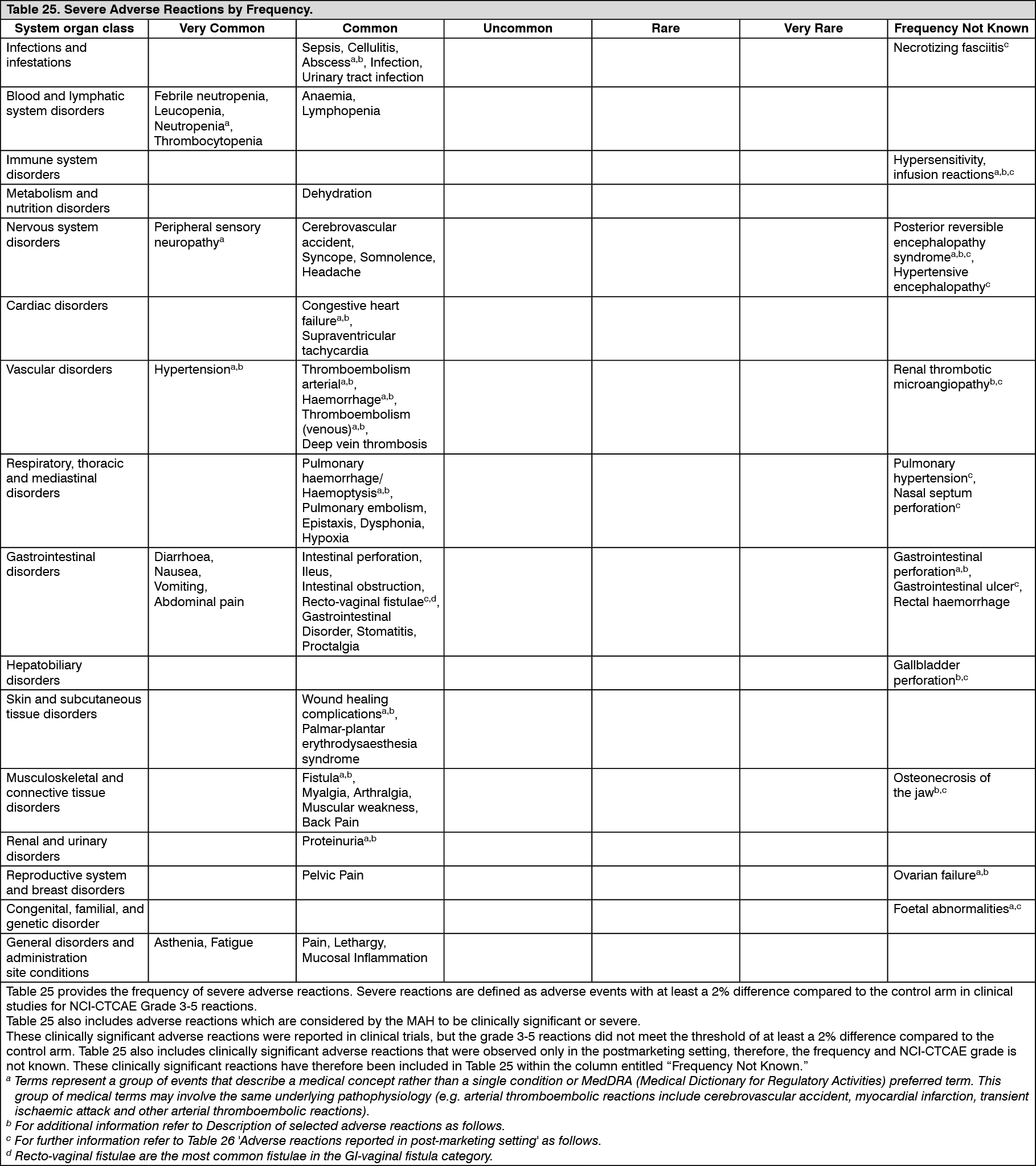 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image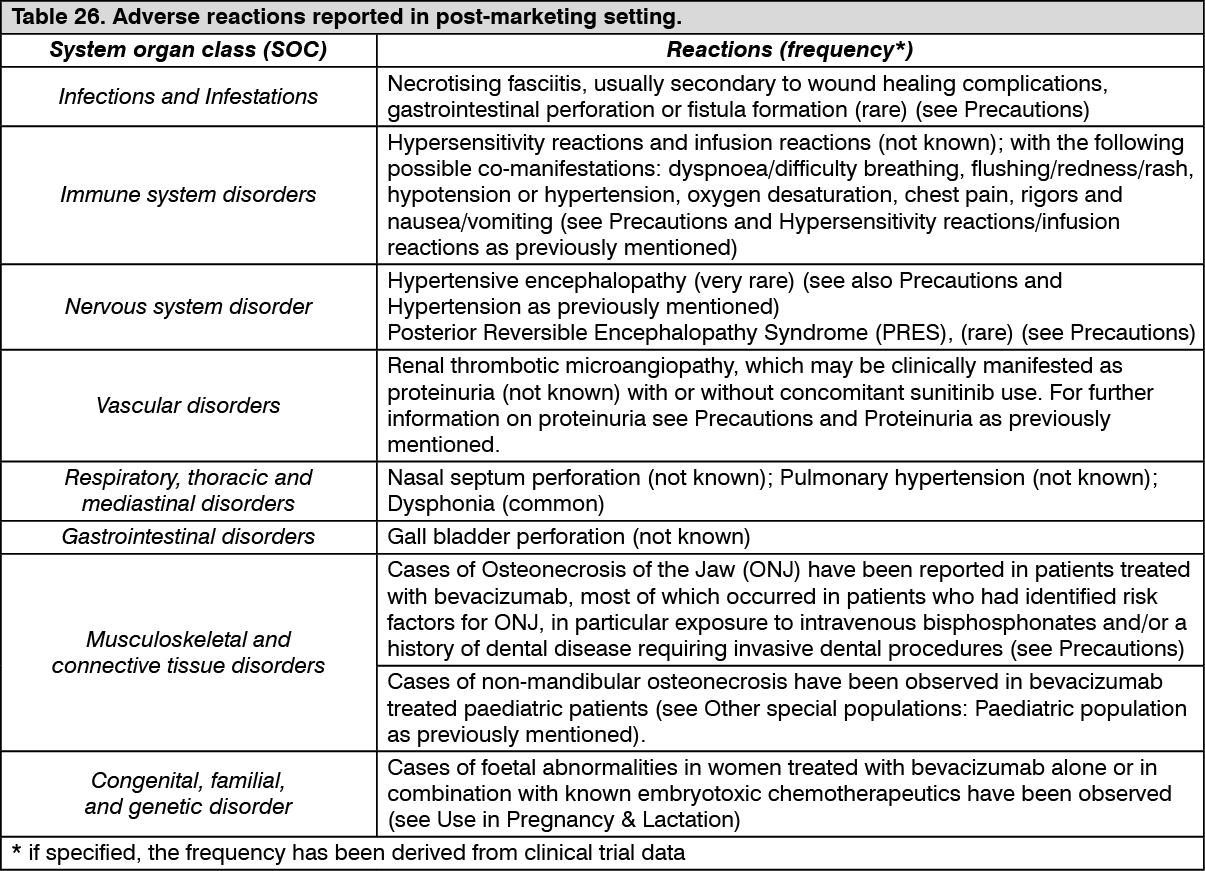 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
