


 Sign Out
Sign Out
Pharmacology: Pharmacodynamics: Mechanism of action and pharmacodynamic effects: Bictegravir is an integrase strand transfer inhibitor (INSTI) that binds to the integrase active site and blocks the strand transfer step of retroviral deoxyribonucleic acid (DNA) integration which is essential for the HIV replication cycle. Bictegravir has activity against HIV-1 and HIV-2.
Emtricitabine is a nucleoside reverse transcriptase inhibitor (NRTI) and analogue of 2'-deoxycytidine. Emtricitabine is phosphorylated by cellular enzymes to form emtricitabine triphosphate. Emtricitabine triphosphate inhibits HIV replication through incorporation into viral DNA by the HIV reverse transcriptase (RT), which results in DNA chain-termination. Emtricitabine has activity against HIV-1, HIV-2 and HBV.
Tenofovir alafenamide is a nucleotide reverse transcriptase inhibitor (NtRTI) and phosphonamidate prodrug of tenofovir (2'-deoxyadenosine monophosphate analogue). Tenofovir alafenamide is permeable into cells and due to increased plasma stability and intracellular activation through hydrolysis by cathepsin A, tenofovir alafenamide is more efficient than tenofovir disoproxil fumarate in loading tenofovir into peripheral blood mononuclear cells (PBMCs) (including lymphocytes and other HIV target cells) and macrophages. Intracellular tenofovir is subsequently phosphorylated to the pharmacologically active metabolite tenofovir diphosphate. Tenofovir diphosphate inhibits HIV replication through incorporation into viral DNA by the HIV RT, which results in DNA chain-termination. Tenofovir has activity against HIV-1, HIV-2 and HBV.
Antiviral activity in vitro: The antiviral activity of bictegravir against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, PBMCs, primary monocyte/macrophage cells, and CD4+ T-lymphocytes. The 50% effective concentration (EC50) values for bictegravir were in the range of < 0.05 to 6.6 nM. The protein-adjusted EC95 of bictegravir was 361 nM (0.162 µg/mL) for wild type HIV-1 virus. Bictegravir displayed antiviral activity in cell culture against HIV-1 group (M, N, O), including subtypes A, B, C, D, E, F, and G (EC50 values ranged from < 0.05 to 1.71 nM), and activity against HIV-2 (EC50 = 1.1 nM).
The antiviral activity of emtricitabine against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, the MAGI CCR5 cell line, and PBMCs. The EC50 values for emtricitabine were in the range of 0.0013 to 0.64 µM. Emtricitabine displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, and G (EC50 values ranged from 0.007 to 0.075 µM) and showed activity against HIV-2 (EC50 values ranged from 0.007 to 1.5 µM).
The antiviral activity of tenofovir alafenamide against laboratory and clinical isolates of HIV-1 subtype B was assessed in lymphoblastoid cell lines, PBMCs, primary monocyte/macrophage cells, and CD4+ T-lymphocytes. The EC50 values for tenofovir alafenamide were in the range of 2.0 to 14.7 nM. Tenofovir alafenamide displayed antiviral activity in cell culture against all HIV-1 groups (M, N, O), including subtypes A, B, C, D, E, F, and G (EC50 values ranged from 0.10 to 12.0 nM) and activity against HIV-2 (EC50 values ranged from 0.91 to 2.63 nM).
Resistance: In vitro: HIV-1 isolates with reduced susceptibility to bictegravir have been selected in cell culture. In one selection, amino acid substitutions M50I and R263K emerged and phenotypic susceptibility to bictegravir was reduced 1.3-, 2.2-, and 2.9-fold for M50I, R263K, and M50I + R263K, respectively. In a second selection, amino acid substitutions T66I and S153F emerged and phenotypic susceptibility to bictegravir was shifted 0.4-, 1.9-, and 0.5-fold for T66I, S153F, and T66I + S153F, respectively.
HIV-1 isolates with reduced susceptibility to emtricitabine have been selected in cell culture and had M184V/I mutations in HIV-1 RT.
HIV-1 isolates with reduced susceptibility to tenofovir alafenamide have been selected in cell culture and had the K65R mutation in HIV-1 RT; in addition, a K70E mutation in HIV-1 RT has been transiently observed. HIV-1 isolates with the K65R mutation have low level reduced susceptibility to abacavir, emtricitabine, tenofovir, and lamivudine. In vitro drug resistance selection studies with tenofovir alafenamide have shown no development of high-level resistance after extended culture.
In treatment-naïve (Studies GS-US-380-1489 and GS-US-380-1490) and virologically-suppressed patients (Studies GS-US-380-1844 and GS-US-380-1878), no patient receiving Biktarvy had HIV-1 with treatment-emergent genotypic or phenotypic resistance to bictegravir, emtricitabine, or tenofovir alafenamide in the final resistance analysis population (n = 10) with HIV-1 RNA ≥ 200 copies/mL at the time of confirmed virologic failure, Week 48, Week 96 (treatment naïve studies only) or early study drug discontinuation. At the time of study entry, one treatment-naïve patient had pre-existing INSTI resistance-associated mutations Q148H + G140S and had HIV-1 RNA < 50 copies/mL at Week 4 through Week 96. In addition, 6 patients had the pre-existing INSTI resistance-associated mutation T97A; all had HIV-1 RNA < 50 copies/mL at Week 96 or the last visit.
Cross-resistance: The susceptibility of bictegravir was tested against 64 INSTI-resistant clinical isolates (20 with single substitutions and 44 with 2 or more substitutions). Of these, all single and double mutant isolates lacking Q148H/K/R and 10 of 24 isolates with Q148H/K/R with additional INSTI resistance associated substitutions had ≤ 2.5-fold reduced susceptibility to bictegravir; > 2.5-fold reduced susceptibility to bictegravir was found for 14 of the 24 isolates that contained G140A/C/S and Q148H/R/K substitutions in integrase. Of those, 9 of the 14 isolates had additional mutations at L74M, T97A, or E138A/K. In a separate study, site-directed mutants with G118R and T97A+G118R had 3.4- and 2.8-fold reduced susceptibility to bictegravir, respectively. The relevance of these in vitro cross-resistance data remains to be established in clinical practice.
Bictegravir demonstrated equivalent antiviral activity against 5 nonnucleoside reverse transcriptase inhibitor (NNRTI)-resistant, 3 NRTI-resistant, and 4 protease inhibitor (PI)-resistant HIV-1 mutant clones compared with the wild-type strain.
Emtricitabine-resistant viruses with the M184V/I substitution were cross-resistant to lamivudine, but retained sensitivity to didanosine, stavudine, tenofovir, and zidovudine.
The K65R and K70E mutations result in reduced susceptibility to abacavir, didanosine, lamivudine, emtricitabine, and tenofovir, but retain sensitivity to zidovudine. Multinucleoside resistant HIV-1 with a T69S double insertion mutation or with a Q151M mutation complex including K65R showed reduced susceptibility to tenofovir alafenamide.
Clinical data: The efficacy and safety of Biktarvy in HIV-1 infected, treatment-naïve adults are based on 48-week and 96-week data from two randomized, double-blind, active-controlled studies, GS-US-380-1489 (n = 629) and GS-US-380-1490 (n = 645).
The efficacy and safety of Biktarvy in virologically-suppressed HIV-1 infected adults are based on 48-week data from a randomised, double-blind, active-controlled study, GS-US-380-1844 (n = 563); and a randomized, open label, active-controlled study, GS-US-380-1878 (n = 577).
HIV-1 infected, treatment-naïve patients: In Study GS-US-380-1489, patients were randomised in a 1:1 ratio to receive either bictegravir/emtricitabine/tenofovir alafenamide (B/F/TAF) (n = 314) or abacavir/dolutegravir/lamivudine (600/50/300 mg) (n = 315) once daily. In Study GS-US-380-1490, patients were randomised in a 1:1 ratio to receive either B/F/TAF (n = 320) or dolutegravir + emtricitabine/tenofovir alafenamide (50+200/25 mg) (n = 325) once daily.
In Studies GS-US-380-1489 and GS-US-380-1490, the mean age was 35 years (range 18-77), 89% were male, 58% were White, 33% were Black, and 3% were Asian. Twenty-four percent of patients identified as Hispanic/Latino. The prevalence of different subtypes was comparable across all three treatment groups, with subtype B predominant in both groups; 11% were non-B subtypes. The mean baseline plasma HIV-1 RNA was 4.4 log10 copies/mL (range 1.3-6.6). The mean baseline CD4+ cell count was 460 cells/mm3 (range 0-1,636) and 11% had CD4+ cell counts less than 200 cells/mm3. Eighteen percent of patients had baseline viral loads greater than 100,000 copies/mL. In both studies, patients were stratified by baseline HIV-1 RNA (less than or equal to 100,000 copies/mL, greater than 100,000 copies/mL to less than or equal to 400,000 copies/mL, or greater than 400,000 copies/mL), by CD4+ cell count (less than 50 cells/μL, 50-199 cells/μL, or greater than or equal to 200 cells/μL), and by region (US or ex-US).
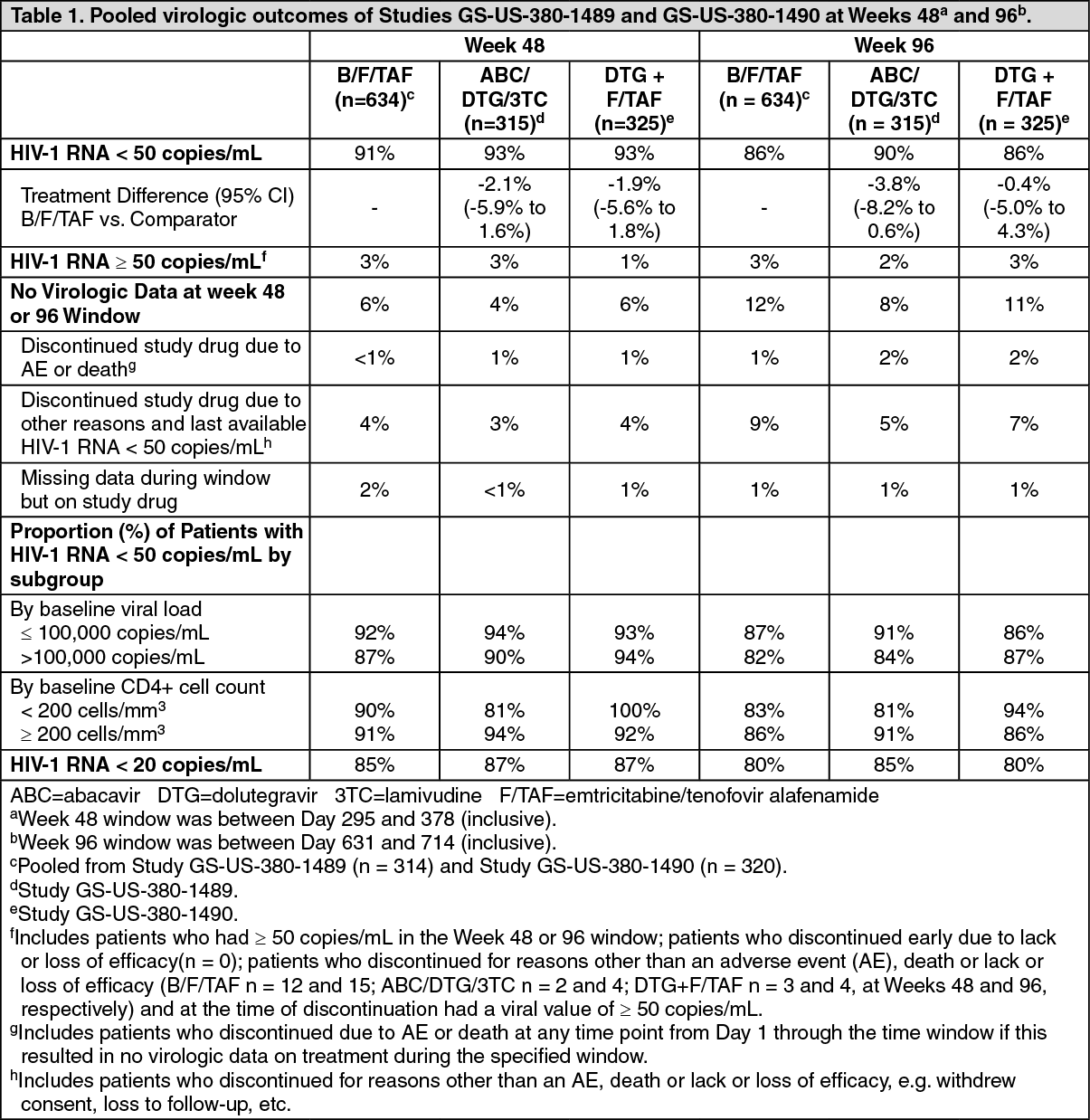
Treatment outcomes of Studies GS-US-380-1489 and GS-US-380-1490 through Weeks 48 and 96 are presented in Table 1. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageB/F/TAF was non-inferior in achieving HIV-1 RNA < 50 copies/mL at both Weeks 48 and 96 when compared to abacavir/dolutegravir/lamivudine and dolutegravir + emtricitabine/tenofovir alafenamide, respectively. Treatment outcomes between treatment groups were similar across subgroups by age, sex, race, baseline viral load, baseline CD4+ cell count, and region.
In Studies GS-US-380-1489 and GS-US-380-1490, the mean increase from baseline in CD4+ cell count at Week 96 was 262, 288, and 281 cells/mm3 in the pooled B/F/TAF, abacavir/dolutegravir/lamivudine, and dolutegravir + emtricitabine/tenofovir alafenamide groups, respectively.
HIV-1 infected, virologically-suppressed patients: In Study GS-US-380-1844, the efficacy and safety of switching from a regimen of dolutegravir+ abacavir/lamivudine or abacavir/dolutegravir/lamivudine to B/F/TAF were evaluated in a randomised, double-blind study of virologically-suppressed (HIV-1 RNA < 50 copies/mL) HIV-1 infected adults (n = 563). Patients must have been stably suppressed (HIV-1 RNA < 50 copies/mL) on their baseline regimen for at least 3 months prior to study entry. Patients were randomised in a 1:1 ratio to either switch to B/F/TAF at baseline (n = 282), or stay on their baseline antiretroviral regimen (n = 281). Patients had a mean age of 45 years (range 20-71), 89% were male, 73% were White, and 22% were Black. Seventeen percent of patients identified as Hispanic/Latino. The prevalence of different HIV-1 subtypes was comparable between treatment groups, with subtype B predominant in both groups; 5% were non-B subtypes. The mean baseline CD4+ cell count was 723 cells/mm3 (range 124-2,444).
In Study GS-US-380-1878, the efficacy and safety of switching from either abacavir/lamivudine or emtricitabine/tenofovir disoproxil fumarate (200/300 mg) plus atazanavir or darunavir (boosted by either cobicistat or ritonavir) to B/F/TAF were evaluated in a randomised, open-label study of virologically-suppressed HIV-1 infected adults (n = 577). Patients must have been stably suppressed on their baseline regimen for at least 6 months and must not have been previously treated with any INSTI. Patients were randomised in a 1:1 ratio to either switch to B/F/TAF (n = 290), or stay on their baseline antiretroviral regimen (n = 287). Patients had a mean age of 46 years (range 20-79), 83% were male, 66% were White, and 26% were Black. Nineteen percent of patients identified as Hispanic/Latino. The mean baseline CD4+ cell count was 663 cells/mm3 (range 62-2,582). The prevalence of different subtypes was comparable across treatment groups, with subtype B predominant in both groups; 11% were non-B subtypes. Patients were stratified by prior treatment regimen. At screening, 15% of patients were receiving abacavir/lamivudine plus atazanavir or darunavir (boosted by either cobicistat or ritonavir) and 85% of patients were receiving emtricitabine/tenofovir disoproxil fumarate plus atazanavir or darunavir (boosted by either cobicistat or ritonavir).
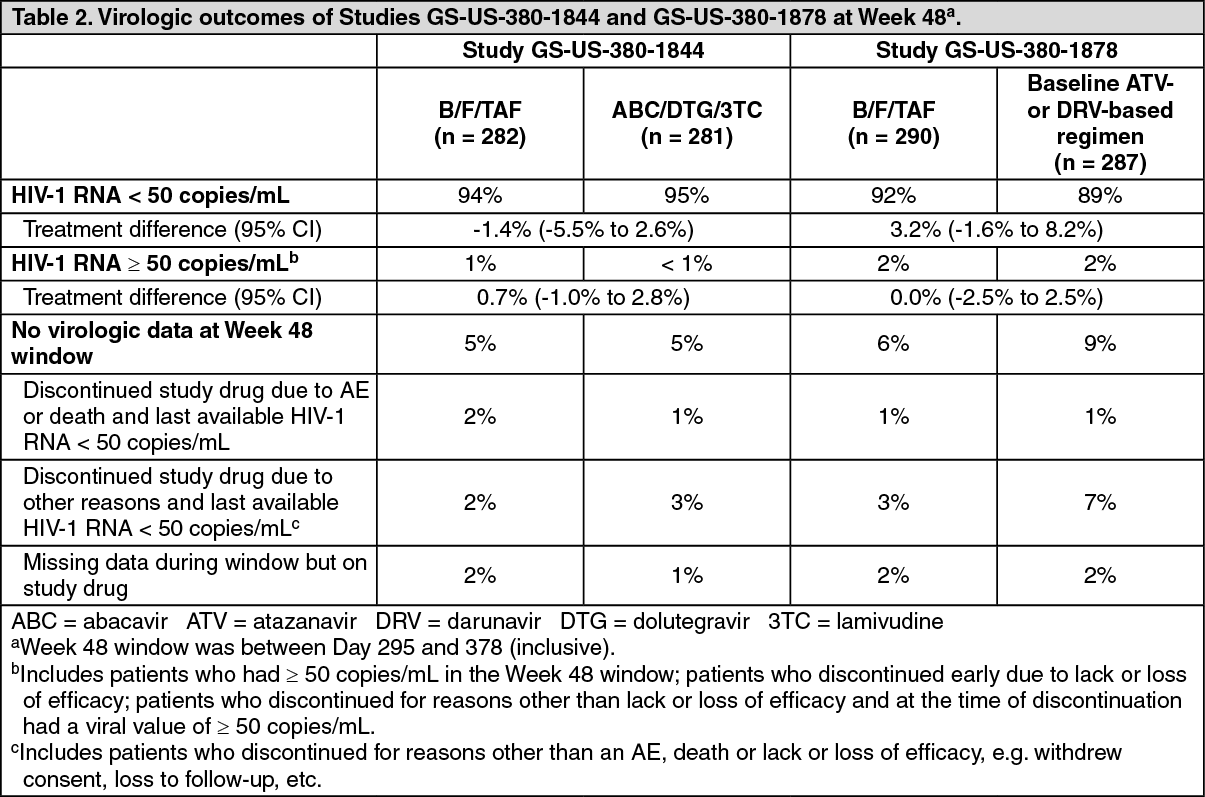
Treatment outcomes of Studies GS-US-380-1844 and GS-US-380-1878 through Week 48 are presented in Table 2. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageB/F/TAF was non-inferior to the control regimen in both studies. Treatment outcomes between treatment groups were similar across subgroups by age, sex, race, and region.
In GS-US-380-1844, the mean change from baseline in CD4+ cell count at Week 48 was - 31 cells/mm3 in patients who switched to B/F/TAF and 4 cells/mm3 in patients who stayed on abacavir/dolutegravir/lamivudine. In GS-US-380-1878, the mean change from baseline in CD4+ cell count at Week 48 was 25 cells/mm3 in patients who switched to B/F/TAF and 0 cells/mm3 in patients who stayed on their baseline regimen.
Patients co-infected with HIV and HBV: The number of patients co-infected with HIV and HBV treated with B/F/TAF is limited. In Study GS-US-380-1490, 7 of 8 patients with HIV/HBV co-infection at baseline who were randomised to receive B/F/TAF. At Week 48, 7 patients were HBV suppressed (HBV DNA < 29 IU/mL) and had HIV-1 RNA < 50 copies/mL. One patient had missing HBV DNA data at Week 48. At Week 96, 4 patients were HBV suppressed and had HIV-1 RNA < 50 copies/mL. Four patients had missing HBV DNA data at Week 96 (one lost to follow-up from Week 48, 1 lost to follow-up after Week 72, 1 with missing HBV data but HIV-1 RNA < 50 copies/mL, and 1 with missing data in Week 96 window).
In Study GS-US-380-1878, at Week 48, 100% (8/8) of the patients co-infected with HIV/HBV at baseline in the B/F/TAF arm maintained HBV DNA < 29 IU/mL (missing = excluded analysis) and HIV RNA < 50 copies/mL.
Pharmacokinetics: Absorption: Bictegravir is absorbed following oral administration with peak plasma concentrations occurring at 2.0-4.0 hours after administration of B/F/TAF. Relative to fasting conditions, the administration of B/F/TAF with either a moderate fat (~600 kcal, 27% fat) or high fat meal (~800 kcal, 50% fat) resulted in an increase in bictegravir AUC (24%). This modest change is not considered clinically meaningful and B/F/TAF can be administered with or without food.
Following oral administration of B/F/TAF with or without food in HIV-1 infected adults, the multiple dose mean (CV%) pharmacokinetic parameters of bictegravir were Cmax = 6.15 µg/mL (22.9%), AUCtau = 102 µg•h/mL (26.9%), and Ctrough = 2.61 µg/mL (35.2%).
Emtricitabine is rapidly and extensively absorbed following oral administration with peak plasma concentrations occurring at 1.5-2.0 hours after administration of B/F/TAF. The mean absolute bioavailability of emtricitabine from 200 mg hard capsules was 93%. Emtricitabine systemic exposure was unaffected when emtricitabine was administered with food and B/F/TAF can be administered with or without food.
Following oral administration of B/F/TAF with or without food in HIV-1 infected adults, the multiple dose mean (CV%) pharmacokinetic parameters of emtricitabine were Cmax = 2.13 µg/mL (34.7%), AUCtau = 12.3 µg•h/mL (29.2%), and Ctrough = 0.096 µg/mL (37.4%).
Tenofovir alafenamide is rapidly absorbed following oral administration with peak plasma concentrations occurring at 0.5-2.0 hours after administration of B/F/TAF. Relative to fasting conditions, the administration of tenofovir alafenamide with a moderate fat meal (~600 kcal, 27% fat) and a high fat meal (~800 kcal, 50% fat) resulted in an increase in AUClast by 48% and 63%, respectively. These modest changes are not considered clinically meaningful and B/F/TAF can be administered with or without food.
Following oral administration of B/F/TAF with or without food in HIV-1 infected adults, the multiple dose mean (CV%) pharmacokinetic parameters of tenofovir alafenamide were Cmax = 0.121 µg/mL (15.4%), and AUCtau = 0.142 µg•h/mL (17.3%).
Distribution: In vitro binding of bictegravir to human plasma proteins was > 99% (free fraction ~0.25%). The in vitro human blood to plasma bictegravir concentration ratio was 0.64.
In vitro binding of emtricitabine to human plasma proteins was < 4% and independent of concentration over the range of 0.02 to 200 µg/mL. At peak plasma concentration, the mean plasma to blood drug concentration ratio was ~1.0 and the mean semen to plasma drug concentration ratio was ~4.0.
In vitro binding of tenofovir to human plasma proteins is < 0.7% and is independent of concentration over the range of 0.01-25 µg/mL. Ex vivo binding of tenofovir alafenamide to human plasma proteins in samples collected during clinical studies was approximately 80%.
Biotransformation: Metabolism is the major clearance pathway for bictegravir in humans. In vitro phenotyping studies showed that bictegravir is primarily metabolised by CYP3A and UGT1A1. Following a single dose oral administration of [14C]-bictegravir, ~60% of the dose from faeces included unchanged parent, desfluoro-hydroxy- BIC-cysteine-conjugate, and other minor oxidative metabolites. Thirty-five percent of the dose was recovered from urine and consisted primarily of the glucuronide of bictegravir and other minor oxidative metabolites and their phase II conjugates. Renal clearance of the unchanged parent was minimal.
Following administration of [14C]-emtricitabine, complete recovery of the emtricitabine dose was achieved in urine (~86%) and faeces (~14%). Thirteen percent of the dose was recovered in the urine as three putative metabolites. The biotransformation of emtricitabine includes oxidation of the thiol moiety to form the 3'-sulfoxide diastereomers (~9% of dose) and conjugation with glucuronic acid to form 2'-O-glucuronide (~4% of dose). No other metabolites were identifiable.
Metabolism is a major elimination pathway for tenofovir alafenamide in humans, accounting for >80% of an oral dose. In vitro studies have shown that tenofovir alafenamide is metabolised to tenofovir (major metabolite) by cathepsin A in PBMCs (including lymphocytes and other HIV target cells) and macrophages; and by carboxylesterase-1 in hepatocytes. In vivo, tenofovir alafenamide is hydrolysed within cells to form tenofovir (major metabolite), which is phosphorylated to the active metabolite, tenofovir diphosphate. In human clinical studies, a 25 mg oral dose of tenofovir alafenamide resulted in tenofovir diphosphate concentrations > 4-fold higher in PBMCs and > 90%lower concentrations of tenofovir in plasma as compared to a 300 mg oral dose of tenofovir disoproxil fumarate.
Elimination: Bictegravir is primarily eliminated by hepatic metabolism. Renal excretion of intact bictegravir is a minor pathway (~1% of dose). The plasma bictegravir half-life was 17.3 hours.
Emtricitabine is primarily excreted by the kidneys by both glomerular filtration and active tubular secretion. The plasma emtricitabine half-life was approximately 10 hours.
Tenofovir alafenamide is eliminated following metabolism to tenofovir. Tenofovir alafenamide and tenofovir have a median plasma half-life of 0.51 and 32.37 hours, respectively. Tenofovir is eliminated by the kidneys by both glomerular filtration and active tubular secretion. Renal excretion of intact tenofovir alafenamide is a minor pathway with less than 1% of the dose eliminated in urine.
Linearity: The multiple dose pharmacokinetics of bictegravir are dose proportional over the dose range of 25 to 100 mg. The multiple dose pharmacokinetics of emtricitabine are dose proportional over the dose range of 25 to 200 mg. Tenofovir alafenamide exposures are dose proportional over the dose range of 8 mg to 125 mg.
Other special populations: Renal impairment: No clinically relevant differences in bictegravir, tenofovir alafenamide, or tenofovir pharmacokinetics were observed between healthy subjects and subjects with severe renal impairment (estimated CrCl < 30 mL/min). There are no pharmacokinetic data on bictegravir or tenofovir alafenamide in patients with creatinine clearance less than 15 mL/min. Mean systemic emtricitabine exposure was higher in patients with severe renal impairment (CrCl < 30 mL/min) (33.7 µg•h/mL) than in subjects with normal renal function (11.8 µg•h/mL).
Hepatic impairment: Clinically relevant changes in the pharmacokinetics of bictegravir were not observed in subjects with moderate hepatic impairment. The pharmacokinetics of emtricitabine have not been studied in subjects with hepatic impairment; however, emtricitabine is not significantly metabolised by liver enzymes, so the impact of liver impairment should be limited. Clinically relevant changes in the pharmacokinetics of tenofovir alafenamide or its metabolite tenofovir were not observed in patients with mild, moderate, or severe hepatic impairment.
Age, gender and race: Pharmacokinetics of bictegravir, emtricitabine, and tenofovir have not been fully evaluated in the elderly (≥ 65 years of age). Population analyses using pooled pharmacokinetic data from adult studies did not identify any clinically relevant differences due to age, gender or race on the exposures of bictegravir, emtricitabine, or tenofovir alafenamide.
Toxicology: Preclinical Safety data: Bictegravir was not mutagenic or clastogenic in conventional genotoxicity assays.
Bictegravir was not carcinogenic in a 6-month rasH2 transgenic mouse study (at doses of up to 100 mg/kg/day in males and 300 mg/kg/day in females, which resulted in exposures of approximately 15 and 23 times, in males and females, respectively, the exposure in humans at the recommended human dose) nor in a 2-year rat study (at doses of up to 300 mg/kg/day, which resulted in exposures of approximately 31 times the exposure in humans).
Studies of bictegravir in monkeys revealed the liver as the primary target organ of toxicity. Hepatobiliary toxicity was described in a 39-week study at a dosage of 1,000 mg/kg/day, which resulted in exposures of approximately 16 times the exposure in humans at the recommended human dose, and was partially reversible after a 4-week recovery period.
Studies in animals with bictegravir have shown no evidence of teratogenicity or an effect on reproductive function. In offspring from rat and rabbit dams treated with bictegravir during pregnancy, there were no toxicologically significant effects on developmental endpoints.
Non-clinical data on emtricitabine reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction and development. Emtricitabine has demonstrated low carcinogenic potential in mice and rats.
Non-clinical studies of tenofovir alafenamide in rats and dogs revealed bone and kidney as the primary target organs of toxicity. Bone toxicity was observed as reduced bone mineral density in rats and dogs at tenofovir exposures at least 43 times greater than those expected after administration of B/F/TAF. A minimal infiltration of histiocytes was present in the eye in dogs at tenofovir alafenamide and tenofovir exposures of approximately 14 and 43 times greater, respectively, than those expected after administration of B/F/TAF.
Tenofovir alafenamide was not mutagenic or clastogenic in conventional genotoxicity assays. Because there is a lower tenofovir exposure in rats and mice after the administration of tenofovir alafenamide compared to tenofovir disoproxil fumarate, carcinogenicity studies and a rat peri-postnatal study were conducted only with tenofovir disoproxil fumarate. No special hazard for humans was revealed in conventional studies of carcinogenic potential and toxicity to reproduction and development. Reproductive toxicity studies in rats and rabbits showed no effects on mating, fertility, pregnancy or foetal parameters. However, tenofovir disoproxil fumarate reduced the viability index and weight of pups in a peri-postnatal toxicity study at maternally toxic doses.