Pharmacotherapeutic group: Vaccines, viral vaccines.
ATC code: J07BN01.
Pharmacology: Pharmacodynamics: Mechanism of action: The nucleoside-modified messenger RNA in Comirnaty is formulated in lipid nanoparticles, which enable delivery of the non-replicating RNA into host cells to direct transient expression of the SARS-CoV-2 Santigen. The mRNA codes for membrane-anchored, full-length S with two-point mutations within the central helix. Mutation of these two amino acids to proline locks S in an antigenically preferred prefusion conformation. The vaccine elicits both neutralising antibody and cellular immune responses to the spike (S) antigen, which may contribute to protection against COVID-19.
Efficacy: Omicron-adapted Comirnaty: Immunogenicity in participants 12 years of age and older - after the booster (fourth dose): In an analysis of a subset from Study 5, 105 participants 12 to 17 years of age, 297 participants 18 through 55 years of age, and 286 participants 56 years of age and older who had previously received a 2-dose primary series and booster dose with Comirnaty received a booster (fourth dose) of Comirnaty Original/Omicron BA.4-5. In participants 12 through 17 years of age, 18 through 55 years of age, and 56 years of age and older, 75.2%, 71.7% and 61.5% were positive for SARS-CoV-2 at baseline, respectively.
Analyses of 50% neutralizing antibody titres (NT50) against Omicron BA.4-5 and against reference strain among participants 56 years of age and older who received a booster (fourth dose) of Comirnaty Original/Omicron BA.4-5 in Study 5 compared to a subset of participants from Study 4 who received a booster (fourth dose) of Comirnaty demonstrated superiority of Comirnaty Original/Omicron BA.4-5 to Comirnaty based on geometric mean ratio (GMR) and noninferiority based on difference in seroresponse rates with respect to anti-Omicron BA.4-5 response, and noninferiority of anti-reference strain immune response based on GMR (Table 1).
Analyses of NT50 against Omicron BA.4/BA.5 among participants 18 through 55 years of age compared to participants 56 years of age and older who received a booster (fourth dose) of Comirnaty Original/Omicron BA.4-5 in Study 5 demonstrated noninferiority of anti-Omicron BA.4-5 response among participants 18 through 55 years of age compared to participants 56 years of age and older for both GMR and difference in seroresponse rates (Table 1).
The study also assessed the level of NT50 of the anti-Omicron BA.4-5 SARS-CoV-2 and reference strains pre-vaccination and 1 month after vaccination in participants who received a booster (fourth dose) (Table 2). (See Tables 1 and 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Comirnaty: Study 2 is a multicentre, multinational, Phase 1/2/3 randomised, placebo-controlled, observer-blind dose-finding, vaccine candidate selection and efficacy study in participants 12 years of age and older. Randomisation was stratified by age: 12 to 15 years of age, 16 to 55 years of age, or 56 years of age and older, with a minimum of 40% of participants in the ≥56-year stratum. The study excluded participants who were immunocompromised and those who had previous clinical or microbiological diagnosis of COVID-19. Participants with pre-existing stable disease, defined as disease not requiring significant change in therapy or hospitalization for worsening disease during the 6 weeks before enrolment, were included as were participants with known stable infection with human immunodeficiency virus (HIV), hepatitis C virus (HCV) or hepatitis B virus (HBV).
Efficacy in participants 16 years of age and older - after 2 doses: In the Phase 2/3 portion of Study 2, based on data accrued through 14 November 2020, approximately 44,000 participants were randomised equally and were to receive 2 doses of the initially approved COVID-19 mRNA Vaccine or placebo. The efficacy analyses included participants that received their second vaccination within 19 to 42 days after their first vaccination. The majority (93.1%) of vaccine recipients received the second dose 19 days to 23 days after Dose 1. Participants are planned to be followed for up to 24 months after Dose 2, for assessments of safety and efficacy against COVID-19. In the clinical study, participants were required to observe a minimum interval of 14 days before and after administration of an influenza vaccine in order to receive either placebo or COVID-19 mRNA Vaccine. In the clinical study, participants were required to observe a minimum interval of 60 days before or after receipt of blood/plasma products or immunoglobulins within through conclusion of the study in order to receive either placebo or COVID-19 mRNA Vaccine.
The population for the analysis of the primary efficacy endpoint included, 36,621 participants 12 years of age and older (18,242 in the COVID-19 mRNA Vaccine group and 18,379 in the placebo group) who did not have evidence of prior infection with SARS-CoV-2 through 7 days after the second dose. In addition, 134 participants were between the ages of 16 to 17 years of age (66 in the COVID-19 mRNA Vaccine group and 68 in the placebo group) and 1,616 participants 75 years of age and older (804 in the COVID-19 mRNA Vaccine group and 812 in the placebo group).
At the time of the primary efficacy analysis, participants had been followed for symptomatic COVID-19 for in total 2,214 person-years for the COVID-19 mRNA Vaccine and in total 2,222 person-years in the placebo group.
There were no meaningful clinical differences in overall vaccine efficacy in participants who were at risk of severe COVID-19 including those with 1 or more comorbidities that increase the risk of severe COVID-19 (e.g., asthma, body mass index (BMI) ≥30 kg/m
2, chronic pulmonary disease, diabetes mellitus, hypertension).
The vaccine efficacy information is presented in Table 3. (See Table 3.)
Click on icon to see table/diagram/image
Efficacy of COVID-19 mRNA Vaccine in preventing first COVID-19 occurrence from 7 days after Dose 2 compared to placebo was 94.6% (95% confidence interval of 89.6% to 97.6%) in participants 16 years of age and older with or without evidence of prior infection with SARS-CoV-2.
Additionally, subgroup analyses of the primary efficacy endpoint showed similar efficacy point estimates across genders, ethnic groups, and participants with medical comorbidities associated with high risk of severe COVID-19.
Updated efficacy analyses were performed with additional confirmed COVID-19 cases accrued during blinded placebo-controlled follow-up, representing up to 6 months after Dose 2 in the efficacy population.
The updated vaccine efficacy information is presented in Table 4. (See Table 4.)
Click on icon to see table/diagram/image
In the updated efficacy analysis, efficacy of COVID-19 mRNA Vaccine in preventing first COVID-19 occurrence from 7 days after Dose 2 compared to placebo was 91.1% (95% CI of 88.8% to 93.0%) during the period when Wuhan/Wild type and Alpha variants were the predominant circulating strains in participants in the evaluable efficacy population with or without evidence of prior infection with SARS-CoV-2.
Additionally, the updated efficacy analyses by subgroup showed similar efficacy point estimates across sexes, ethnic groups, geography and participants with medical comorbidities and obesity associated with high risk of severe COVID-19.
Efficacy against severe COVID-19: Updated efficacy analyses of secondary efficacy endpoints supported benefit of the COVID-19 mRNA Vaccine in preventing severe COVID‑19.
As of 13 March 2021, vaccine efficacy against severe COVID-19 is presented only for participants with or without prior SARS-CoV-2 infection (Table 5) as the COVID-19 case counts in participants without prior SARS-CoV-2 infection were the same as those in participants with or without prior SARS-CoV-2 infection in both the COVID-19 mRNA Vaccine and placebo groups. (See Table 5.)
Click on icon to see table/diagram/image
Efficacy and immunogenicity in adolescents 12 to 15 years of age - after 2 doses: In an initial analysis of Study 2 in adolescents 12 to 15 years of age (representing a median follow-up duration of >2 months after Dose 2) without evidence of prior infection, there were no cases in 1,005 participants who received the vaccine and 16 cases out of 978 who received placebo. The point estimate for efficacy is 100% (95% confidence interval 75.3, 100.0). In participants with or without evidence of prior infection there were 0 cases in the 1,119 who received vaccine and 18 cases in 1,110 participants who received placebo. This also indicates the point estimate for efficacy is 100% (95% confidence interval 78.1, 100.0).
Updated efficacy analyses were performed with additional confirmed COVID-19 cases accrued during blinded placebo-controlled follow-up, representing up to 6 months after Dose 2 in the efficacy population.
In the updated efficacy analysis of Study 2 in adolescents 12 to 15 years of age without evidence of prior infection, there were no cases in 1,057 participants who received the vaccine and 28 cases out of 1,030 who received placebo. The point estimate for efficacy is 100% (95% confidence interval 86.8, 100.0) during the period when Alpha variant was the predominant circulating strain. In participants with or without evidence of prior infection there were 0 cases in the 1,119 who received vaccine and 30 cases in 1,109 participants who received placebo. This also indicates the point estimate for efficacy is 100% (95% confidence interval 87.5, 100.0).
In Study 2, an analysis of SARS-CoV-2 neutralising titres 1 month after Dose 2 was conducted in a randomly selected subset of participants who had no serological or virological evidence of past SARS-CoV-2 infection up to 1 month after Dose 2, comparing the response in adolescents 12 to 15 years of age (n=190) to participants 16 to 25 years of age (n=170).
The ratio of the geometric mean titres (GMT) in the 12 to 15 years of age group to the 16 to 25 years of age group was 1.76, with a 2-sided 95% CI of 1.47 to 2.10. Therefore, the 1.5-fold noninferiority criterion was met as the lower bound of the 2-sided 95% CI for the geometric mean ratio [GMR] was >0.67.
10 mcg: Immunogenicity in children 5 to 11 years of age (i.e., 5 to less than 12 years of age) - after the booster (fourth dose): In an analysis of a subset from Study 6, 103 participants 5 to 11 years of age who had previously received a 2-dose primary series and booster dose with Comirnaty received a booster (fourth dose) of Comirnaty Original/Omicron BA.4-5. Results include immunogenicity data from a comparator subset of participants 5 to 11 years of age in Study 3 who received 3 doses of Comirnaty. In participants 5 to 11 years of age who received a fourth dose of Comirnaty Original/Omicron BA.4-5 and participants 5 to 11 years of age who received a third dose of Comirnaty, 57.3% and 58.4% were positive for SARS-CoV-2 at baseline, respectively.
The immune response 1 month after a booster dose (fourth dose), Comirnaty Original/Omicron BA.4-5 elicited generally similar Omicron BA.4/BA.5-specific neutralizing titres compared with the titres in the comparator group who received 3 doses of Comirnaty. Comirnaty Original/Omicron BA.4-5 also elicited similar reference strain-specific titres compared with the titres in the comparator group.
The vaccine immunogenicity results after a booster dose in participants 5 to 11 years of age are presented in Table 6. (See Table 6.)
Click on icon to see table/diagram/image
Efficacy and immunogenicity in children 5 to 11 years of age (i.e., 5 to less than 12 years of age) - after 2 doses: Study 3 is a Phase 1/2/3 study comprised of an open-label vaccine dose-finding portion (Phase 1) and a multicentre, multinational, randomised, saline placebo-controlled, observer-blind efficacy portion (Phase 2/3) that has enrolled participants 5 to 11 years of age. The majority (94.4%) of randomised vaccine recipients received the second dose 19 days to 23 days after Dose 1.
Initial descriptive vaccine efficacy results in children 5 to 11 years of age without evidence of prior SARS-CoV-2 infection are presented in Table 7. No cases of COVID-19 were observed in either the vaccine group or the placebo group in participants with evidence of prior SARS-CoV-2 infection. (See Table 7.)
Click on icon to see table/diagram/image
Pre-specified hypothesis-driven efficacy analysis was performed with additional confirmed COVID-19 cases accrued during blinded placebo-controlled follow-up, representing up to 6 months after Dose 2 in the efficacy population.
In the efficacy analysis of Study 3 in children 5 to 11 years of age without evidence of prior infection, there were 10 cases in 2,703 participants who received the vaccine and 42 cases out of 1,348 who received placebo. The point estimate for efficacy is 88.2% (95% confidence interval 76.2, 94.7) during the period when Delta variant was the predominant circulating strain. In participants with or without evidence of prior infection there were 12 cases in the 3,018 who received vaccine and 42 cases in 1,511 participants who received placebo. The point estimate for efficacy is 85.7% (95% confidence interval 72.4, 93.2).
In Study 3, an analysis of SARS-CoV-2 50% neutralising titres (NT50) 1 month after Dose 2 in a randomly selected subset of participants demonstrated effectiveness by immunobridging of immune responses comparing children 5 to 11 years of age (i.e., 5 to less than 12 years of age) in the Phase 2/3 part of Study 3 to participants 16 to 25 years of age in the Phase 2/3 part of Study 2 who had no serological or virological evidence of past SARS CoV-2 infection up to 1 month after Dose 2, meeting the prespecified immunobridging criteria for both the geometric mean ratio (GMR) and the seroresponse difference with seroresponse defined as achieving at least 4-fold rise in SARS-CoV-2 NT50 from baseline (before Dose 1).
The GMR of the SARS-CoV-2 NT50 1 month after Dose 2 in children 5 to 11 years of age (i.e., 5 to less than 12 years of age) to that of young adults 16 to 25 years of age was 1.04 (2-sided 95% CI: 0.93, 1.18). Among participants without prior evidence of SARS-CoV-2 infection up to 1 month after Dose 2, 99.2% of children 5 to 11 years of age and 99.2% of participants 16 to 25 years of age had a seroresponse at 1 month after Dose 2. The difference in proportions of participants who had seroresponse between the 2 age groups (children - young adult) was 0.0% (2-sided 95% CI: -2.0%, 2.2%). This information is presented in Table 8. (See Table 8.)
Click on icon to see table/diagram/image
Immunogenicity in children 5 to 11 years of age (i.e. 5 to less than 12 years of age) - after booster dose: A booster dose of Comirnaty was given to 401 randomly selected participants in Study 3. Effectiveness of a booster dose in ages 5 to 11 is inferred by immunogenicity. The immunogenicity of this was assessed through NT50 against the reference strain of SARS-CoV-2 (USA_WA1/2020). Analyses of NT50 1 month after the booster dose compared to before the booster dose demonstrated a substantial increase in GMTs in individuals 5 through 11 years of age who had no serological or virological evidence of past SARS-CoV-2 infection up to 1 month after the dose 2 and the booster dose. This analysis is summarized in Table 9. (See Table 9.)
Click on icon to see table/diagram/image
30 mcg: Immunogenicity in participants 18 years of age and older - after booster dose: Effectiveness of a booster dose of Comirnaty was based on an assessment of 50% neutralising antibody titres (NT50) against SARS-CoV-2 (USA_WA1/2020) in Study 2. In this study, the booster dose was administered 5 to 8 months (median 7 months) after the second dose. In Study 2, analyses of NT50 1 month after the booster dose compared to 1 month after the primary series in individuals 18 through 55 years of age who had no serological or virological evidence of past SARS-CoV-2 infection up to 1 month after the booster vaccination demonstrated noninferiority for both geometric mean ratio (GMR) and difference in seroresponse rates. Seroresponse for a participant was defined as achieving a ≥4-fold rise in NT50 from baseline (before primary series). These analyses are summarized in Table 10. (See Table 10.)
Click on icon to see table/diagram/image
Relative vaccine efficacy in participants 16 years of age and older - after booster dose: An interim efficacy analysis of Study 4, a placebo-controlled booster study performed in approximately 10,000 participants 16 years of age and older who were recruited from Study 2, evaluated confirmed COVID-19 cases accrued from at least 7 days after booster vaccination up to a data cut-off date of 5 October 2021, which represents a median of 2.5 months post-booster follow-up. The booster dose was administered 5 to 13 months (median 11 months) after the second dose. Vaccine efficacy of the Comirnaty booster dose after the primary series relative to the placebo booster group who only received the primary series dose was assessed.
The relative vaccine efficacy information for participants 16 years of age and older without prior evidence of SARS-CoV-2 infection is presented in Table 11. Relative vaccine efficacy in participants with or without evidence of prior SARS-CoV-2 infection was 94.6% (95% confidence interval of 88.5% to 97.9%), similar to that seen in those participants without evidence of prior infection. Primary COVID-19 cases observed from 7 days after booster vaccination were 7 primary cases in the Comirnaty group, and 124 primary cases in the placebo group. (See Table 11.)
Click on icon to see table/diagram/image
Immunogenicity of a booster dose following primary vaccination with another authorised COVID-19 vaccine: Effectiveness of a Comirnaty booster dose (30 mcg) in individuals who completed primary vaccination with another authorised COVID-19 vaccine (heterologous booster dose) is inferred from immunogenicity data from an independent National Institutes of Health (NIH) study phase ½ open-label clinical trial (NCT04889209) conducted in the United States. In this study, adults (range 19 to 80 years of age) who had completed primary vaccination with Moderna 100 mcg 2-dose series (N=51, mean age 54±17), Janssen single dose (N=53, mean age 48±14), or Comirnaty 30 mcg 2-dose series (N=50, mean age 50±18) at least 12 weeks prior to enrolment and who reported no history of SARS-CoV-2 infection received a booster dose of Comirnaty (30 mcg). The boost with Comirnaty induced a 36, 12, and 20 GMR-fold rise in neutralising titres following the Janssen, Moderna, and Comirnaty primary doses, respectively.
Heterologous boosting with Comirnaty was also evaluated in the CoV-BOOST study (EudraCT 2021-002175-19), a multicentre, randomised, controlled, phase 2 trial of third dose booster vaccination against COVID-19, in which 107 adult participants (median age 71 years of age, interquartile range 54 to 77 years of age) were randomised at least 70 days post 2 doses of AstraZeneca COVID-19 Vaccine. After the AstraZeneca COVID‑19 Vaccine primary series, pseudovirus (wild-type), neutralising antibody NT50 GMR-fold change increased 21.6-fold with heterologous Comirnaty booster (n=95).
Immunogenicity in participants >55 years of age - after a booster dose (fourth dose) of Comirnaty (30 mcg): In an interim analysis of a subset from Study 4 (Substudy E), 305 participants >55 years of age who had completed a series of 3 doses of Comirnaty received Comirnaty (30 mcg) as a booster dose (fourth dose) 5 to 12 months after receiving Dose 3. For the Immunogenicity subset data see Table 12.
Immunogenicity in participants 18 to ≤55 years of age - after a booster dose (fourth dose) of Comirnaty (30 mcg): In Substudy D [a subset from Study 2 (Phase 3) and Study 4 (Phase 3)], 325 participants 18 to ≤55 years of age who had completed 3 doses of Comirnaty received Comirnaty (30 mcg) as a booster dose (fourth dose) 90 to 180 days after receiving Dose 3. For the Immunogenicity subset data see Table 12. (See Table 12.)
Click on icon to see table/diagram/image
Pharmacokinetics: Not applicable.
Toxicology: Preclinical Safety Data: Non-clinical data reveal no special hazard for humans based on conventional studies of repeat dose toxicity and reproductive and developmental toxicity.
General toxicity: Rats intramuscularly administered Comirnaty (receiving 3 full human doses once weekly, generating relatively higher levels in rats due to body weight differences) demonstrated some injection site oedema and erythema and increases in white blood cells (including basophils and eosinophils) consistent with an inflammatory response as well as vacuolation of portal hepatocytes without evidence of liver injury. All effects were reversible.
Genotoxicity/Carcinogenicity: Neither genotoxicity nor carcinogenicity studies were performed. The components of the vaccine (lipids and mRNA) are not expected to have genotoxic potential.
Reproductive toxicity: Reproductive and developmental toxicity were investigated in rats in a combined fertility and developmental toxicity study where female rats were intramuscularly administered Comirnaty prior to mating and during gestation (receiving 4 full human doses that generate relatively higher levels in rat due to body weight differences, spanning between pre-mating day 21 and gestational day 20). SARS-CoV-2 neutralising antibody responses were present in maternal animals from prior to mating to the end of the study on postnatal day 21 as well as in foetuses and offspring. There were no vaccine-related effects on female fertility, pregnancy, or embryo-foetal or offspring development. No Comirnaty data are available on vaccine placental transfer or excretion in milk.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image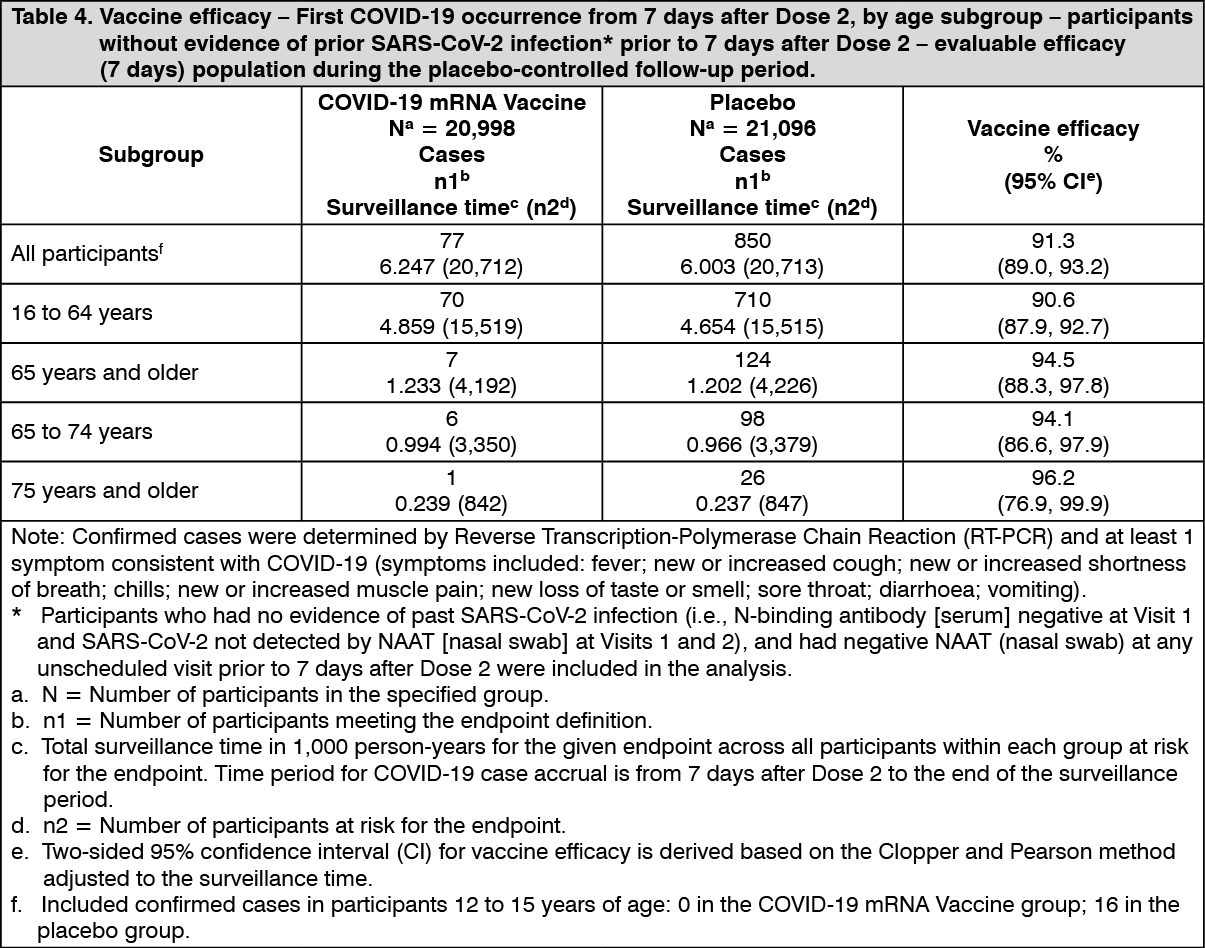 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image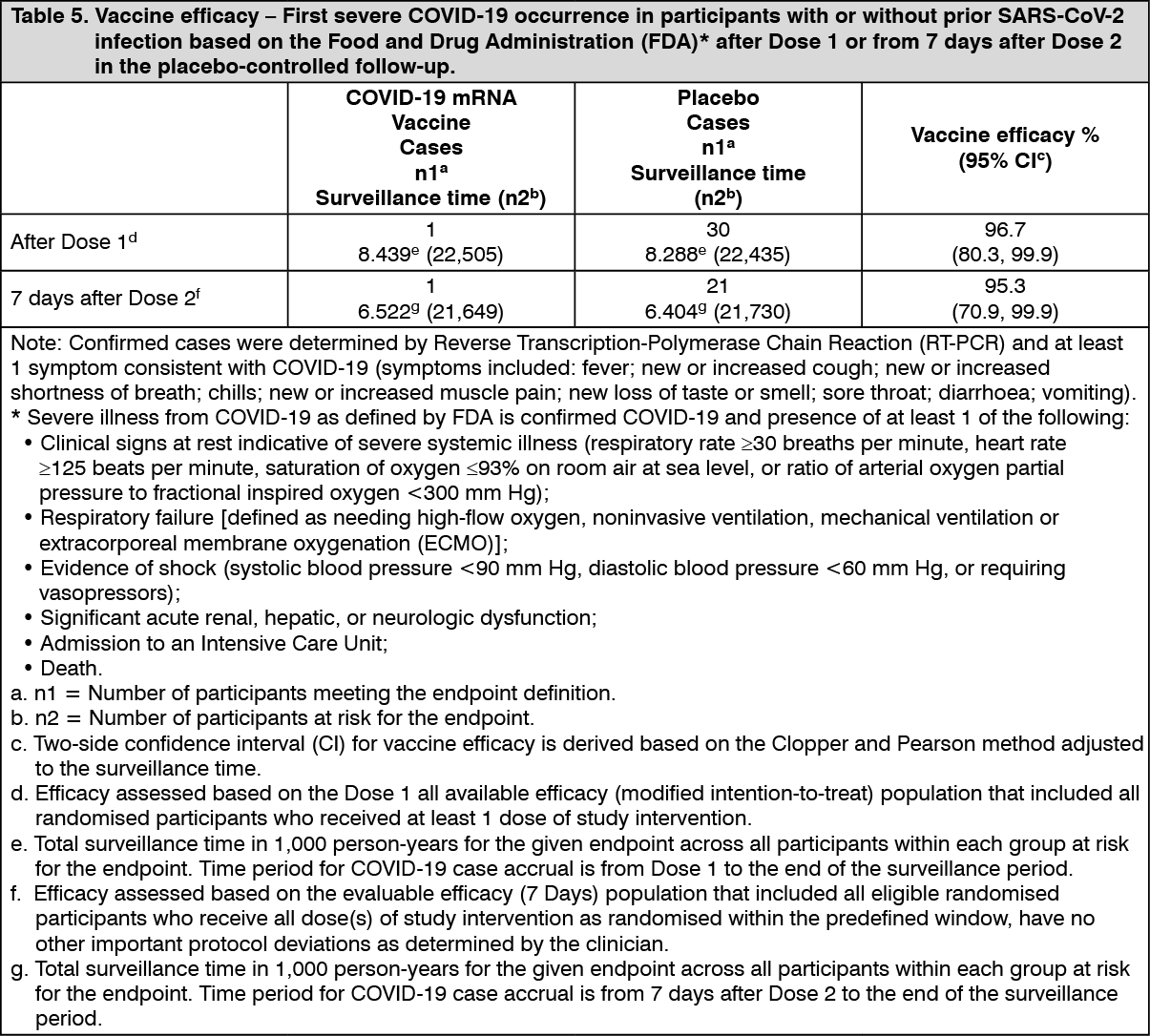 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image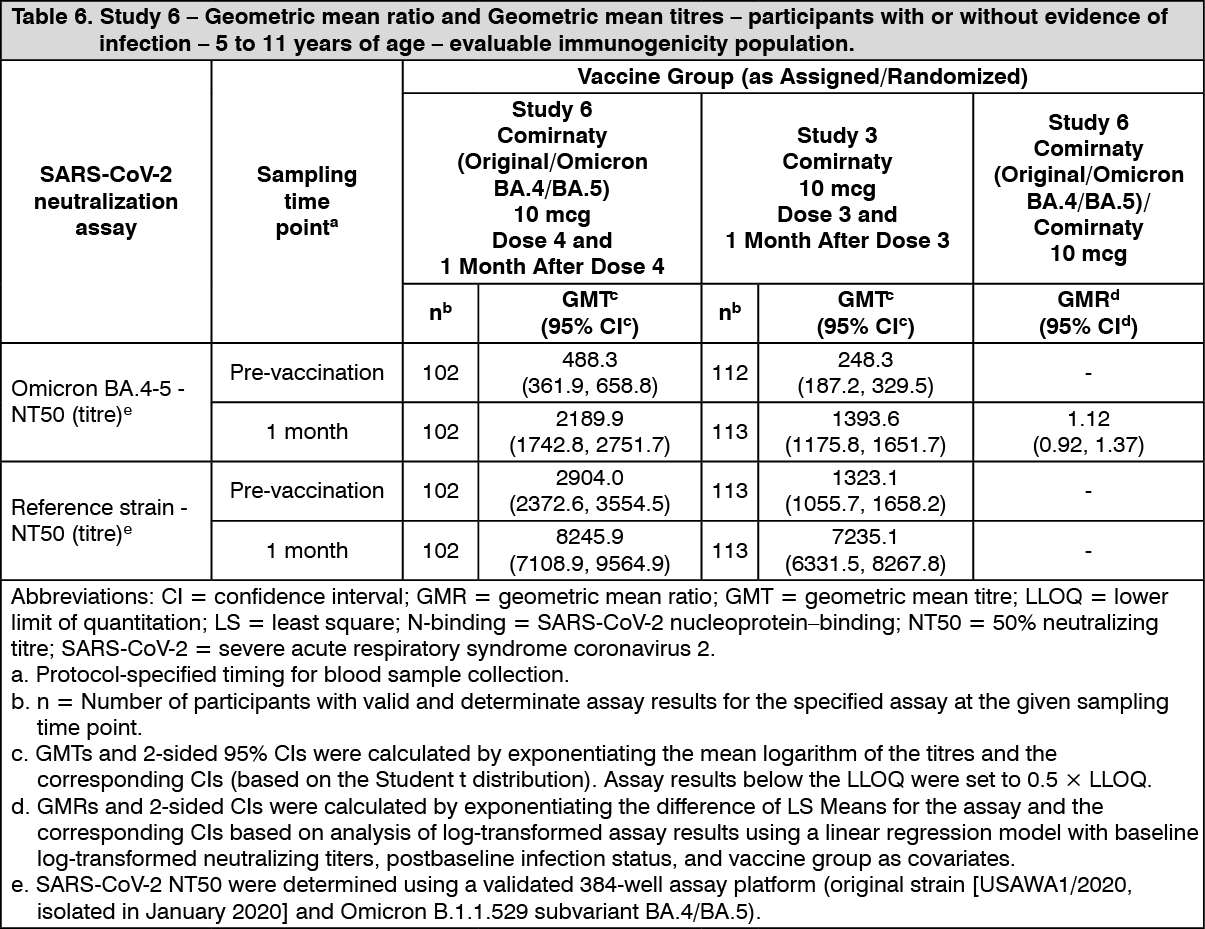 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image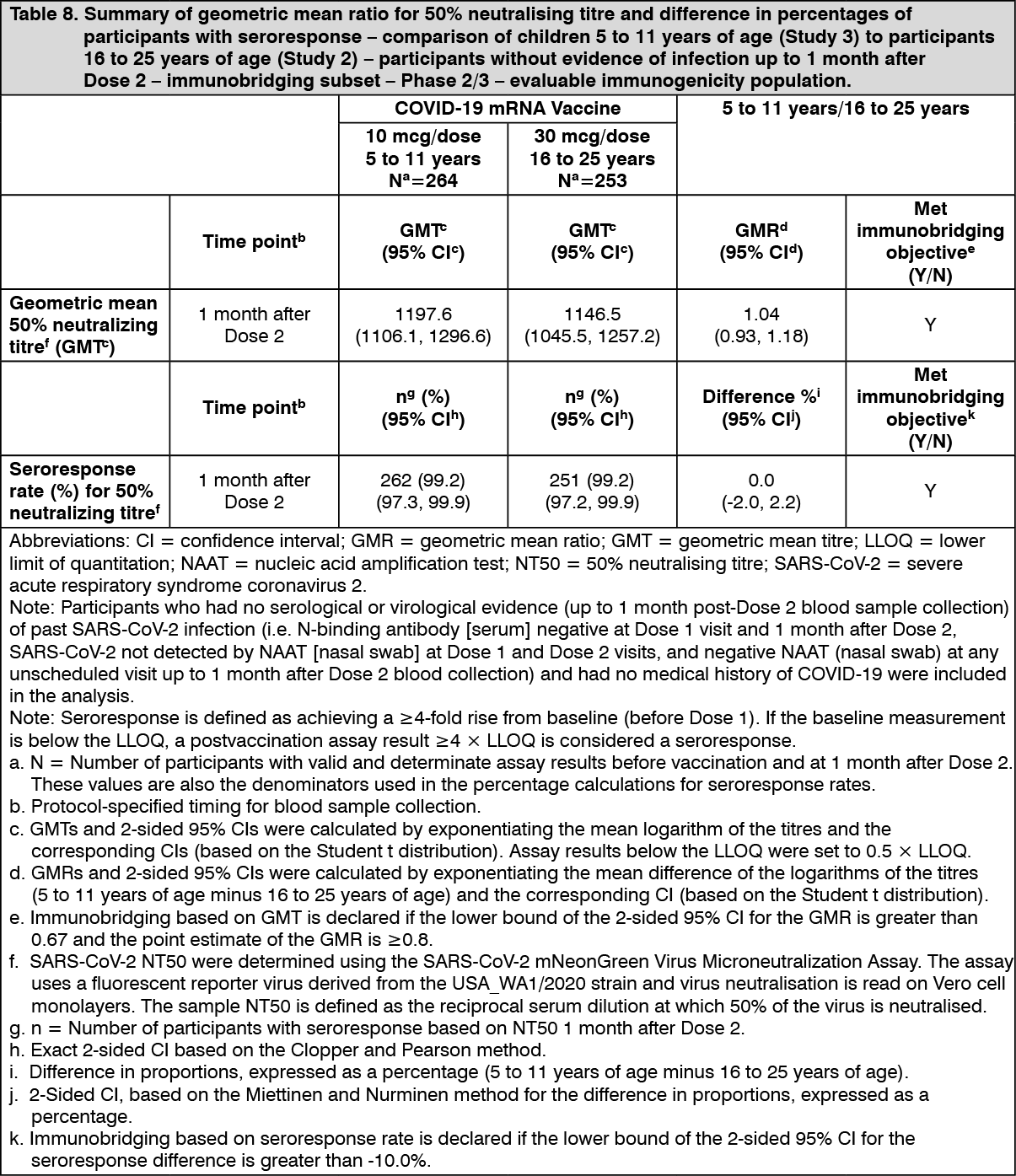 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image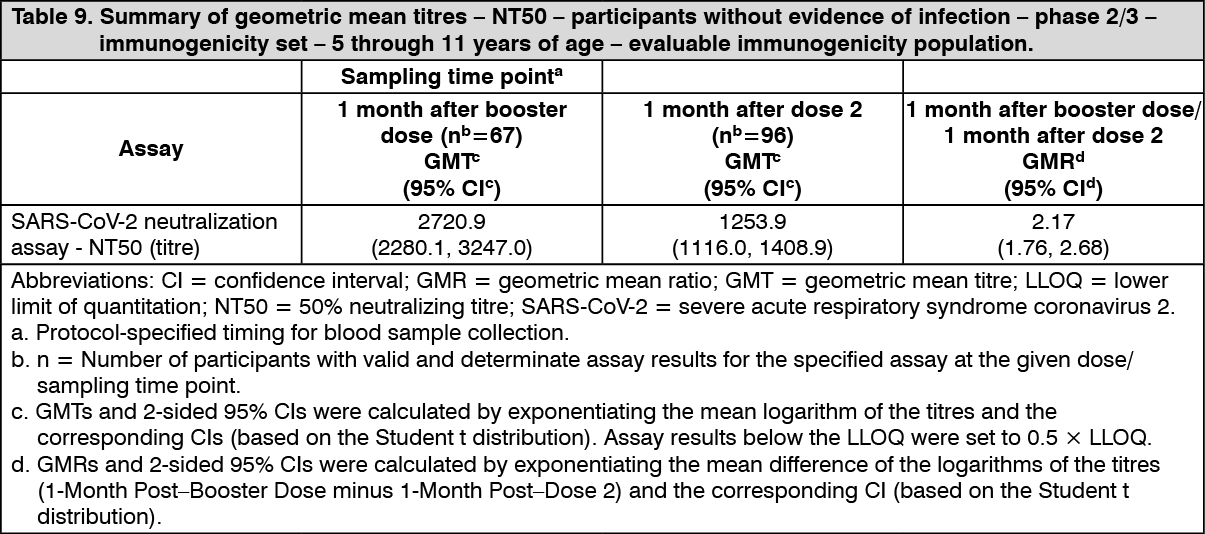 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image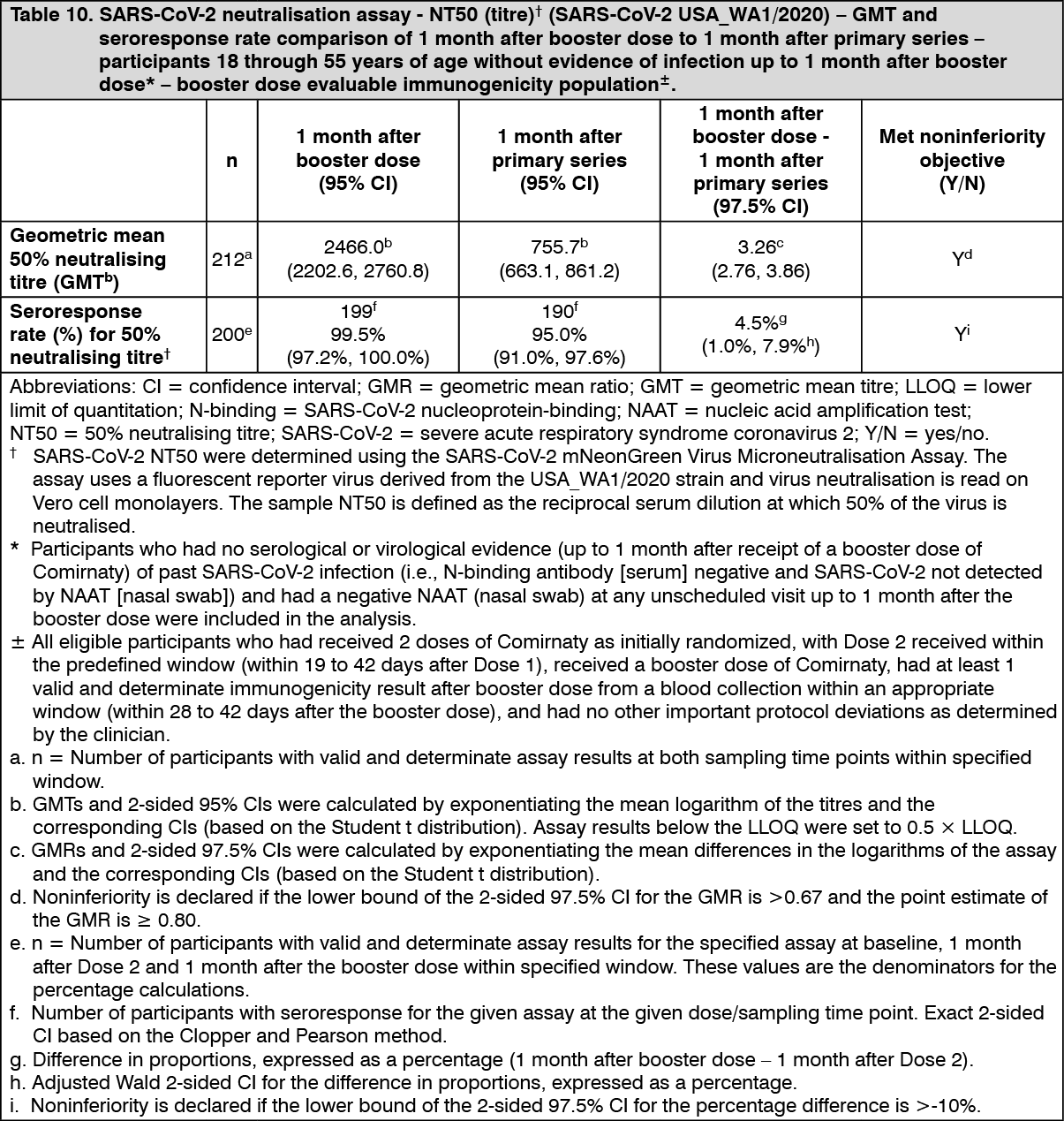 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image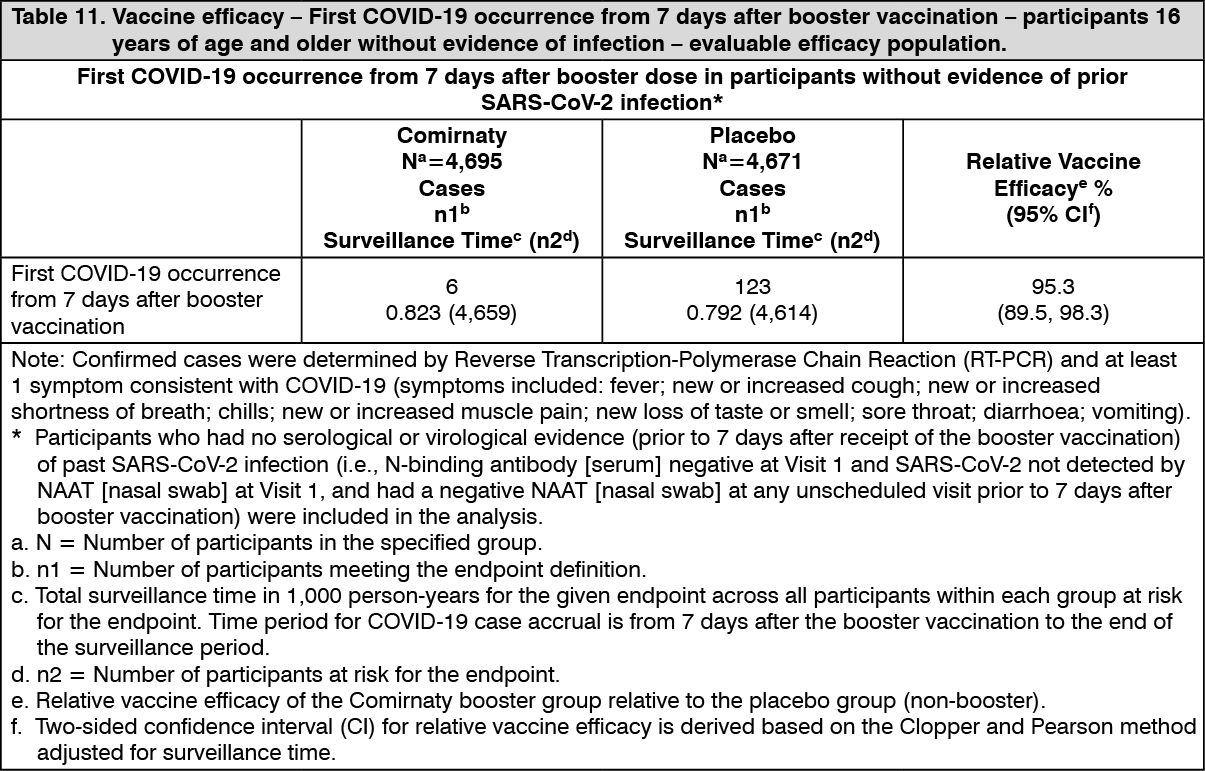 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image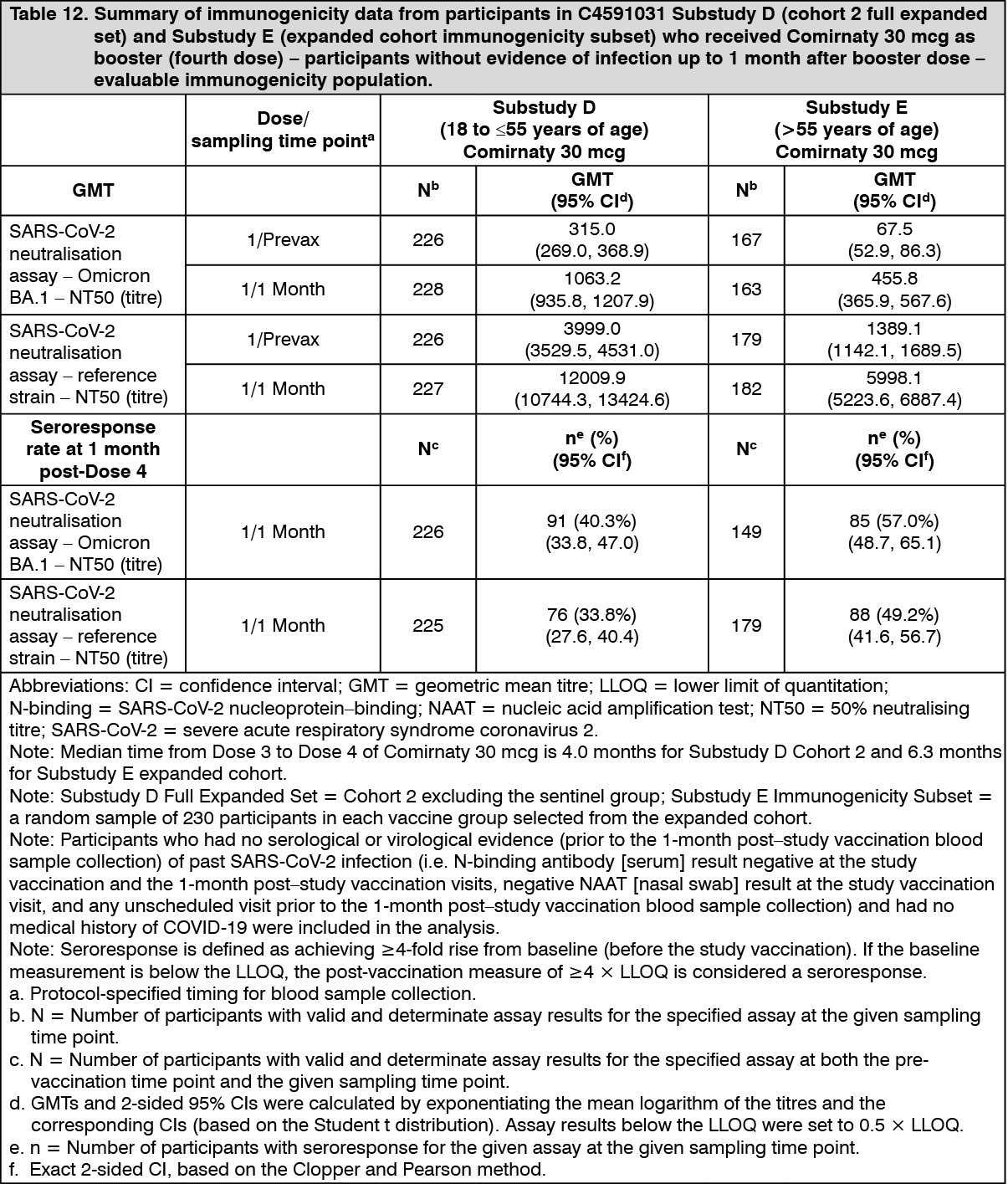 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image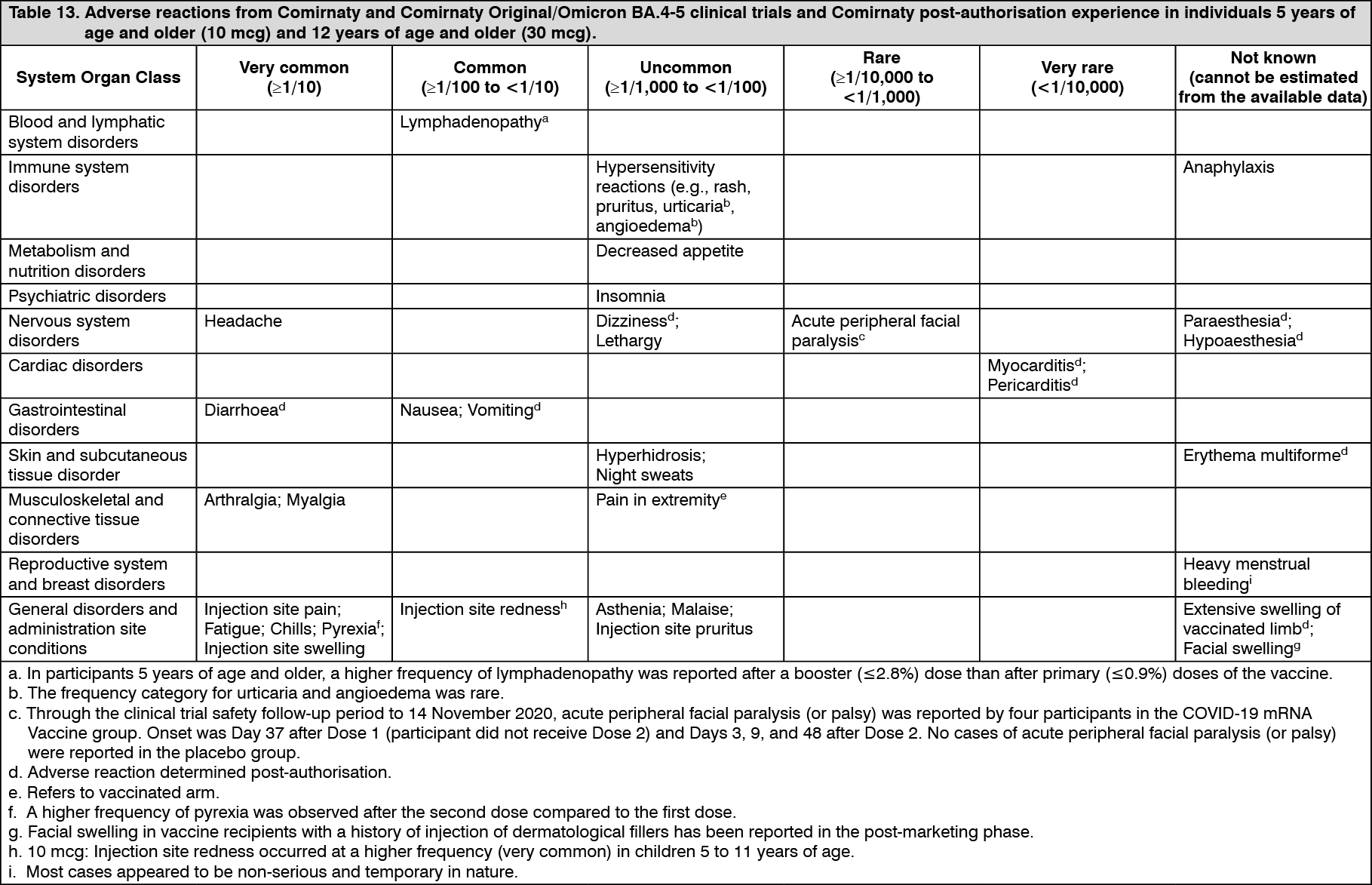 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
