Summary of the safety profile: The most commonly reported adverse reactions in patients treated with CYMBALTA were nausea, dry mouth and headache. However, the majority of common adverse reactions were mild to moderate, they usually started early in therapy, and most tended to subside even as therapy was continued.
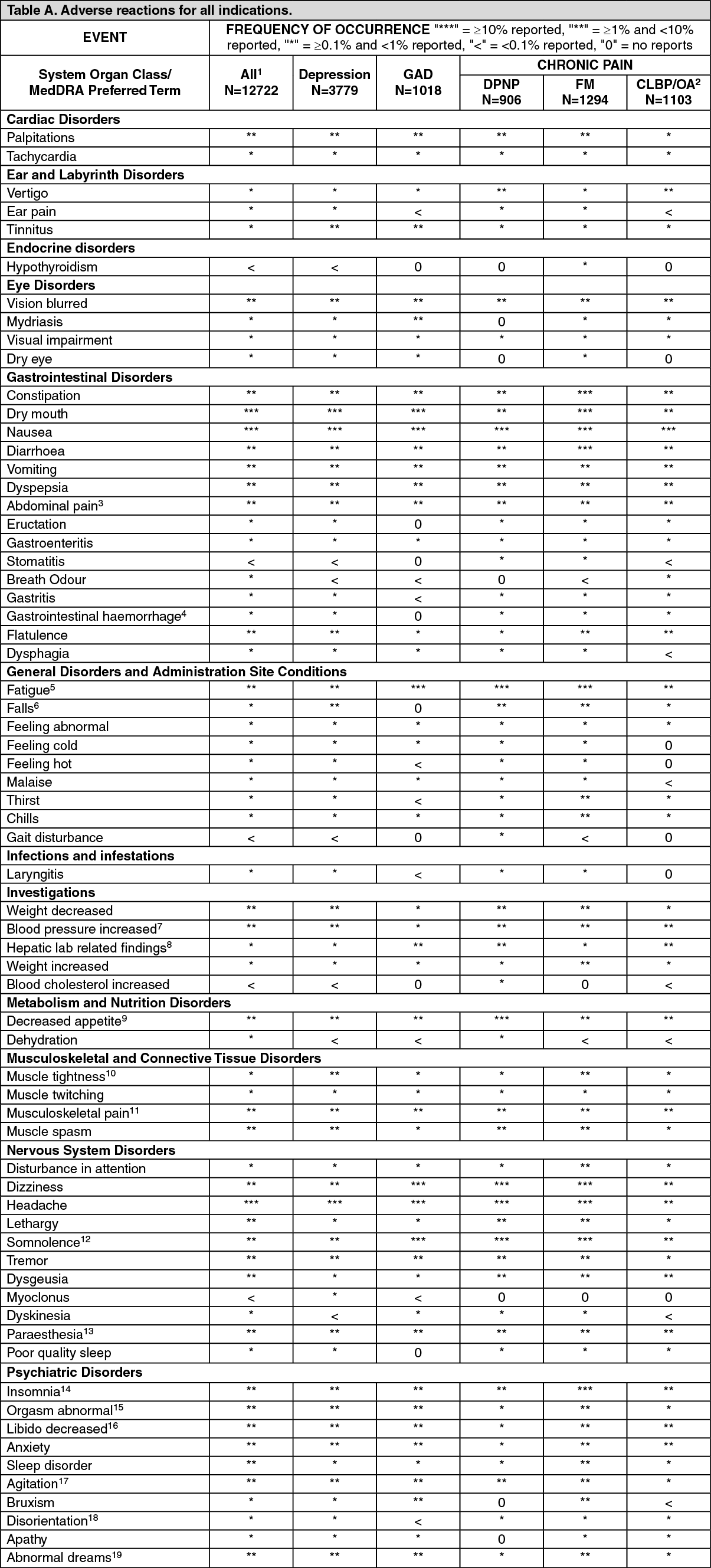
Tabulated summary of adverse reactions: The tables give the adverse reactions observed from clinical trial data for all indication. (See Tables A and B.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
SPONTANEOUS DATA: Adverse drug reactions for all indications:
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
SPONTANEOUS DATA: Adverse drug reactions for all indications: The following list of undesirable effects (adverse drug reactions) is based on post-marketing spontaneous reports, and corresponding reporting rates have been provided.
Cardiac disorders: Very rarely (< 0.01%):
Supraventricular arrhythmia.
Ear and Labyrinth Disorders: Very rarely (< 0.01%):
Tinnitus upon treatment discontinuation.
Endocrine disorders: Very rarely (< 0.01%):
Syndrome of Inappropriate Antidiuretic Hormone (SIADH).
Eye disorders: Very rarely (< 0.01%):
Glaucoma.
Gastrointestinal Disorders: Very rarely (< 0.01%):
Microscopic colitis.
Hepatobiliary disorders: Very rarely (< 0.01%): Hepatitis, jaundice.
Immune system disorders: Very rarely (< 0.01%):
Anaphylactic reaction, hypersensitivity.
Investigations: Very rarely (< 0.01%): Alanine aminotransferase increased, alkaline phosphatase increased, aspartate aminotransferase increased, bilirubin increased.
Metabolism and nutrition disorders: Very rarely (< 0.01%): Hyponatremia, hyperglycemia (reported especially in diabetic patients).
Musculoskeletal and connective tissue disorders: Very rarely (< 0.01%):
Trismus.
Nervous system disorders: Very rarely (< 0.01%): Extrapyramidal disorder, paraesthesia (including electric shock-like sensation) upon treatment discontinuation, restless legs syndrome, serotonin syndrome, seizures, seizures upon treatment discontinuation.
Psychiatric disorders: Rarely (≥ 0.01% -<0.1%): Hallucinations.
Very rarely (< 0.01%):
Mania, aggression and anger, (particularly early in treatment or after treatment discontinuation).
Renal and urinary disorders: Rarely (≥ 0.01% -<0.1%): Urinary retention.
Reproductive system and breast disorders: Very rarely (< 0.01%): Gynecological bleeding, galactorrhea, hyperprolactinemia.
Skin and subcutaneous tissue disorders: Rarely (≥ 0.01% -<0.1%):
Rash.
Very rarely (< 0.01%): Angioneurotic edema, contusion, cutaneous vasculitis (sometimes associated with systemic involvement), ecchymosis, Stevens-Johnson Syndrome, urticarial.
Vascular disorders: Very rarely (< 0.01%):
Orthostatic hypotension (especially at the initiation of treatment), syncope (especially at initiation of treatment), hypertensive crisis.
Description of selected adverse reactions: Discontinuation symptoms have been reported when stopping duloxetine. The most commonly reported symptoms following abrupt or tapered discontinuation of duloxetine in clinical trials have include dizziness, nausea, headache, paresthesia, fatigue, vomiting, irritability, nightmares, insomnia, diarrhea, anxiety, hyperhidrosis, vertigo, somnolence and myalgia.
Generally, for SSRIs and SNRIs, these events are mild to moderate and self-limiting, however, in some patients they may be severe and/or prolonged. It is therefore advised that when duloxetine treatment is no longer required, gradual discontinuation by dose tapering should be carried out (see Dosage & Administration and Precautions).
Duloxetine treatment in placebo-controlled clinical trials was associated with small mean increases from baseline to endpoint in ALT, AST, CPK, and potassium; infrequent, transient, abnormal values were observed for these analytes in duloxetine-treated patients, compared with placebo-treated patients.
Glucose Regulation: In three clinical trials of duloxetine for the treatment of diabetic peripheral neuropathic pain, the mean duration of diabetes was approximately 12 years, the mean baseline fasting blood glucose was 176 mg/dL, and the mean baseline hemoglobin A
1c (HbA
1c) was 7.81%. In the 12 week acute phase of three clinical trials of duloxetine in patients with diabetic neuropathic pain, small but statistically significant increases in fasting blood glucose were observed in duloxetine-treated patients. HbA1c was stable in both duloxetine-treated and placebo-treated patients. In the extension phase of these studies, which lasted up to 52 weeks, there was an increase in HbA1c in both the duloxetine and routine care groups, but the mean increase was 0.3% greater in the duloxetine-treated group. There was also a small increase in fasting blood glucose and in total cholesterol in duloxetine-treated patients while those laboratory tests showed a slight decrease in the routine care group.
Electrocardiograms were obtained from 1139 duloxetine treated patients and 777 placebo-treated patients in 8-week clinical trials in major depressive disorder, and from 528 duloxetine-treated and 205 placebo-treated patients with diabetic neuropathic pain in clinical trials lasting up to 13-weeks. The heart rate-corrected QT interval in duloxetine-treated patients did not differ from that seen in placebo-treated patients. No clinically significant differences were observed for QT, PR, QRS, or QTcB measurements between duloxetine-treated and placebo-treated patients.
Effect on Blood Pressure: Cymbalta treatment, for up to 9-weeks in MDD placebo-controlled clinical trials of 40 to 120 mg daily doses caused increases in blood pressure, averaging 2 mm Hg systolic and 0.5 mm Hg diastolic compared to placebo and an increase in the incidence of at least one measurement of systolic blood pressure over 140 mm Hg.
Cymbalta treatment, for up to 9-weeks in MDD placebo-controlled clinical trials and for up to 13-weeks in DPN placebo-controlled trials caused a small increase in heart rate compared to placebo of about 2 beats per minute.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image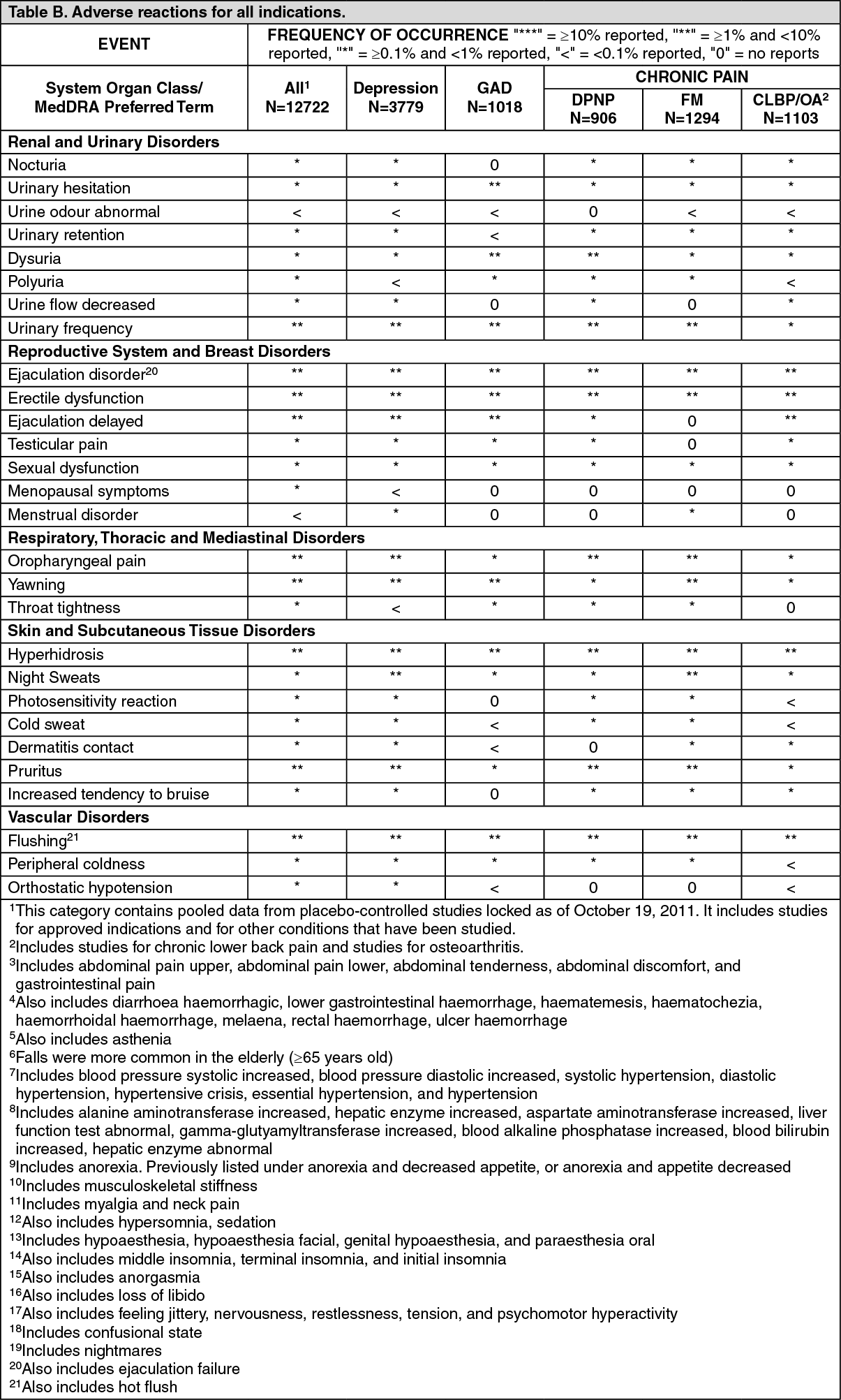 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
