Pharmacology: Pharmacodynamics: Apixaban, a selective inhibitor of the coagulation factor Xa (FXa), is chemically described as 1-(4-Methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5,6,7-tetrahydro-1
H-pyrazolo[3,4-
c]pyridine-3-carboxamide. Its molecular formula is C
25H
25N
5O
4, which corresponds to a molecular weight of 459.5.
Apixaban is a white to pale yellow powder. At physiological pH (1.2-6.8), apixaban does not ionize; its aqueous solubility across the physiological pH range is ~0.04 mg/mL.
The pharmacodynamic effects of apixaban are reflective of the mechanism of action (FXa inhibition). As a result of FXa inhibition, apixaban prolongs clotting tests such as prothrombin time (PT), INR and activated partial thromboplastin time (aPTT). Changes observed in these clotting tests at the expected therapeutic dose are small and subject to a high degree of variability. They are not recommended to assess the pharmacodynamic effects of apixaban. In the thrombin time generation assay, apixaban reduced endogenous thrombin potential, a measure of thrombin generation in human plasma.
Apixaban also demonstrates anti-FXa activity as evident by reduction in FXa enzyme activity in the Rotachrom Heparin chromogenic assay data from clinical studies. Anti-FXa activity exhibits a close direct linear relationship with apixaban plasma concentration, reaching maximum values at the time of apixaban peak plasma concentrations. The relationship between apixaban plasma concentration and anti-FXa activity is linear over a wide dose range of apixaban. The dose- and concentration-related changes observed following apixaban administration are more pronounced, and less variable, with anti-FXa activity compared with clotting tests.
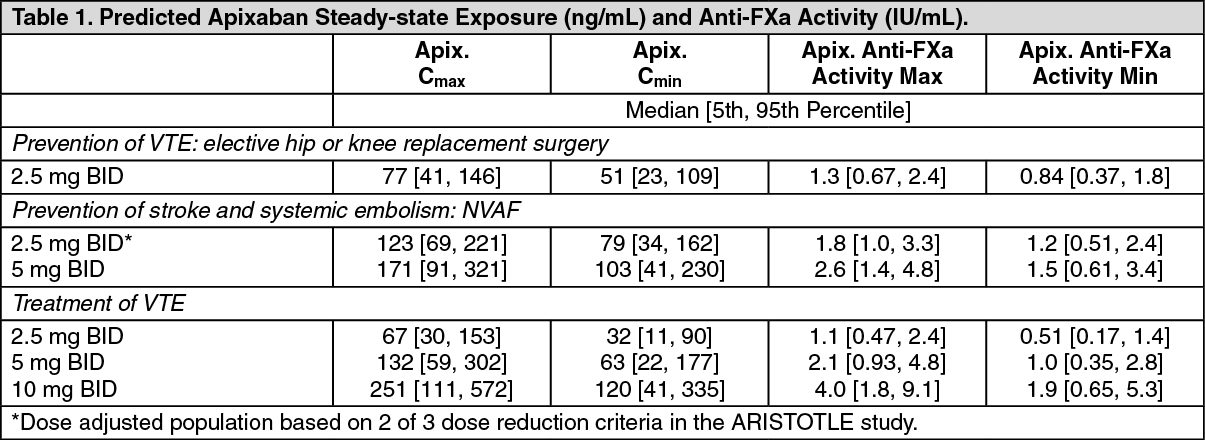
Table 1 shows the predicted steady-state exposure and anti-Factor Xa activity for each indication. In patients taking apixaban for the prevention of VTE following hip or knee replacement surgery, the results demonstrate a less than 1.6-fold fluctuation in peak-to-trough levels. In non-valvular atrial fibrillation patients taking apixaban for the prevention of stroke and systemic embolism, the results demonstrate a less than 1.7-fold fluctuation in peak-to-trough levels. In patients taking apixaban for the treatment of VTE or prevention of recurrence of VTE, the results demonstrate a less than 2.2-fold fluctuation in peak-to-trough levels. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Although treatment with apixaban does not require routine monitoring of exposure, a calibrated quantitative anti-FXa assay may be useful in situations where knowledge of apixaban exposure may help to inform clinical decisions.
Mechanism of Action: Apixaban is a potent, oral, reversible, direct and highly selective active site inhibitor of FXa. It does not require antithrombin III for antithrombotic activity. Apixaban inhibits free and clot-bound FXa, and prothrombinase activity. Apixaban has no direct effects on platelet aggregation, but indirectly inhibits platelet aggregation induced by thrombin. By inhibiting FXa, apixaban prevents thrombin generation and thrombus development. Preclinical studies of apixaban in animal models have demonstrated antithrombotic efficacy in the prevention of arterial and venous thrombosis at doses that preserved hemostasis.
Clinical Studies: Clinical Trial Information:
Prevention of VTE: Elective Hip or Knee Replacement Surgery: The apixaban clinical program was designed to demonstrate the efficacy and safety of apixaban for the prevention of VTE in a broad range of adult patients undergoing elective hip or knee replacement. A total of 8464 patients were randomized in two pivotal, double-blind, multi-national studies, comparing apixaban 2.5 mg given orally twice daily or enoxaparin 40 mg once daily. Included in this total were 1262 patients of age 75 or older, 1004 patients with low body weight (≤60 kg), 1495 patients with BMI ≥33 kg/m
2 and 437 patients with severe or moderate renal impairment. The ADVANCE-3 study included 5407 patients undergoing elective hip replacement, and the ADVANCE-2 study included 3057 patients undergoing elective knee replacement. Subjects received either apixaban 2.5 mg given orally twice daily (po bid) or enoxaparin 40 mg administered subcutaneously once daily (sc od). The first dose of apixaban was given 12 to 24 hours post-surgery, whereas enoxaparin was started 9 to 15 hours prior to surgery. Both apixaban and enoxaparin were given for 32-38 days in the ADVANCE-3 study and for 10 14 days in the ADVANCE-2 study.
Apixaban demonstrated a statistically superior reduction in the primary endpoint, a composite of all VTE/all cause death, and in the Major VTE endpoint, a composite of proximal DVT, non-fatal PE, and VTE-related death, compared to enoxaparin in both elective hip or knee replacement surgery (see Table 2).
Click on icon to see table/diagram/image
The safety endpoints of major bleeding, the composite of major and clinically relevant non-major (CRNM) bleeding, and all bleeding showed similar rates for patients treated with apixaban 2.5 mg compared with enoxaparin 40 mg (see Table 3). All the bleeding criteria included surgical site bleeding.
In both Phase III studies, bleeding was assessed beginning with the first dose of double-blind study drug, which was either enoxaparin or injectable placebo, given 9 to 15 hours before surgery. Bleeding during the treatment period included events that occurred before the first dose of apixaban, which was given 12-24 hours after surgery. Bleeding during the post-surgery treatment period only included events occurring after the first dose of study drug after surgery. Over half the occurrences of major bleeding in the apixaban group occurred prior to the first dose of apixaban. Table 3 shows the bleeding results from the treatment period and the post-surgery treatment period. (See Table 3.)
Click on icon to see table/diagram/image
Prevention of Stroke and Systemic Embolism: NVAF: The clinical program was designed to demonstrate the efficacy and safety of apixaban for the prevention of stroke and systemic embolism in patients suitable for VKA (ARISTOTLE) and in patients unsuitable for VKA (AVERROES). Both studies were active-controlled (vs warfarin in ARISTOTLE and vs ASA in AVERROES), randomized, double-blind, parallel-arm, multi-national trials in patients with nonvalvular, persistent, paroxysmal, or permanent atrial fibrillation (AF) or atrial flutter (AFl) and one or more of the following additional risk factors: Prior stroke or transient ischemic attack (TIA) (also prior systemic embolism in ARISTOTLE), age ≥75 years, arterial hypertension requiring treatment, diabetes mellitus, heart failure New York Heart Association Class 2, decreased left ventricular ejection fraction (LVEF), documented peripheral arterial disease (AVERROES only). (See Table 4.)
Click on icon to see table/diagram/image
ARISTOTLE Study: Patients were randomized to treatment with apixaban 5 mg orally twice daily (or 2.5 mg twice daily in selected patients, 4.7%) or warfarin (target INR range 2.0-3.0) and followed for a median of 89.86 weeks for apixaban and 87.79 weeks for warfarin. The apixaban 2.5 mg twice daily dose was assigned to patients with at least 2 of the following characteristics: Age ≥80 years, body weight ≤60 kg, or serum creatinine ≥1.5 mg/dL (133 micromole/L). 43% were VKA naive, defined as not previously received or have received ≤30 consecutive days of treatment with warfarin or another VKA. Coronary artery disease was present in 33.2% of patients.
For patients randomized to warfarin, the median percentage of time in therapeutic range (INR 2-3) was 66%.
The primary objective of the study was to determine if apixaban 5 mg twice daily (or 2.5 mg twice daily in selected patients) was noninferior to warfarin for the prevention of stroke (ischemic, hemorrhagic, or unspecified) and systemic embolism. Assessments of superiority of apixaban versus warfarin were also prespecified for the primary endpoint and for death due to any cause.
The key study outcomes were prespecified and tested in a sequential, hierarchical manner to conserve overall type 1 error. Apixaban was tested compared to warfarin for: (1) non-inferiority on the composite endpoint of stroke and systemic embolism, (2) superiority on the composite endpoint of stroke and systemic embolism, (3) superiority on major bleeding, and (4) superiority on all-cause death.
In the study, apixaban achieved statistically significant superiority in the primary endpoint of prevention of stroke (hemorrhagic or ischemic) and systemic embolism (see Table 5 and Figure 1). Statistically significant superiority was also achieved in all-cause death (see Table 5), Numeric reductions were observed for both CV and non-CV deaths.
Apixaban reduced the incidence of strokes compared to warfarin within each stroke severity category, including less severe strokes [Rankin score 0 to 2, HR=0.89 (CI=0.64, 1.26)] and the more clinically important fatal or disabling strokes [Rankin score 3 to 6, HR=0.71 (CI=0.54, 0.94)]. The reduction in the stroke and systemic embolism incidence was seen regardless of the stroke risk at entry as categorized by CHADS
2 score. (See Table 5.)
Click on icon to see table/diagram/image
The rate of myocardial infarction was similar between the apixaban and warfarin treatment groups (0.53%/year and 0.61%/year, respectively). (See Figure 1.)
Click on icon to see table/diagram/image
ARISTOTLE study: Centers were ranked
post hoc by the percentage of time that warfarin-treated patients were in therapeutic range (INR 2-3). Findings for stroke/systemic embolism, major bleeds, and all-cause mortality are shown for centers above and below the median level of INR control in Table 6. The benefits of apixaban relative to warfarin were consistent in patients enrolled at centers with INR control below or above the median. (See Table 6.)
Click on icon to see table/diagram/image
AVERROES Study: Patients were randomized to treatment with apixaban 5 mg orally twice daily (or 2.5 mg twice daily in selected patients, 6.4%) or ASA 81 to 324 mg once daily. The selection of an ASA dose of 81, 162, 243, or 324 mg was at the discretion of the investigator with 90.5% of subjects receiving either an 81 mg (64.3%) or 162 mg (26.2%) dose at randomization.
In the study, VKA therapy had been tried but discontinued in 40% of patients prior to enrollment. Common reasons for unsuitability for VKA therapy in the AVERROES study included unable/unlikely to obtain INRs at requested intervals (42.6%), patient refused treatment with VKA (37.4%), CHADS
2 score=1 and physician did not recommend VKA (21.3%), patient could not be relied on to adhere to VKA medication instruction (15.0%), and difficulty/expected difficulty in contacting patient in case of urgent dose change (11.7%).
The primary objective of the study was to determine if apixaban 5 mg twice daily (2.5 mg twice daily in selected patients) was superior to ASA (81-324 mg QD) for preventing the composite outcome of stroke or systemic embolism. Assessments of superiority of apixaban versus ASA were also pre-specified for major vascular events (composite outcome of stroke, systemic embolism, myocardial infarction or vascular death) and for death due to any cause.
AVERROES was stopped early upon the recommendation of the trial's independent Data Monitoring Committee which found that a predefined interim analysis revealed clear evidence of apixaban providing a clinically important reduction in stroke and systemic embolism and acceptable safety profile.
In the study, apixaban demonstrated statistically significant superiority in the primary endpoint of prevention of stroke (hemorrhagic or ischemic) and systemic embolism (see Table 7 and Figure 2). A clinically important reduction was observed in the key secondary composite endpoint of stroke, systemic embolism, myocardial infarction, or vascular death (see Table 7).
Apixaban reduced the incidence of strokes compared to ASA within each stroke severity category [(modified Rankin score 0 to 2, HR=0.51 (CI=0.29, 0.91); modified Rankin score 3 to 6, HR=0.43 (CI=0.28, 0.65)]. The reduction in the stroke incidence was seen regardless of the stroke risk at entry as categorized by CHADS
2 score.
Apixaban also reduced the incidence of cardiovascular hospitalizations relative to ASA (HR =0.79, CI=0.69, 0.91). (See Table 7 and Figure 2.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Bleeding in Patients with Atrial Fibrillation: In the ARISTOTLE and AVERROES studies, the primary safety endpoint was major bleeding, which was defined as acute clinically overt bleeding that was accompanied by one or more of the following: A decrease in hemoglobin of 2 g/dL or more; a transfusion of 2 or more units of packed red blood cells; bleeding that occurred in at least one of the following critical sites, intracranial, intraspinal, intraocular (within the corpus of the eye; thus, a conjunctival bleed is not an intraocular bleed), pericardial, intra-articular, intramuscular with compartment syndrome, retroperitoneal; bleeding that is fatal. Intracranial hemorrhage included intracerebral (including hemorrhagic stroke), subarachnoid, and subdural bleeds.
Clinically relevant non-major bleeding (CRNM) was defined as acute clinically overt bleeding that does not satisfy additional criteria required for the bleeding event to be defined as a major bleeding event and meets at least one of the following criteria: Hospital admission for bleeding; physician guided medical or surgical treatment for bleeding; change in antithrombotic treatment (anticoagulant or antiplatelet) therapy.
ARISTOTLE Study: There was a statistically superior reduction in the incidence of ISTH major bleeding between the apixaban and warfarin treatment groups (see Table 8). There was also a substantial reduction in the incidence of ISTH major+CRNM and all bleeding. (See Table 8.)
Click on icon to see table/diagram/image
Intracranial hemorrhage was reduced >50% with apixaban. GUSTO severe and TIMI major bleeding were reduced >40% with apixaban. Fatal bleeding was reduced >70% with apixaban.
Treatment discontinuation due to bleeding related adverse reactions occurred in 1.7% and 2.5% of patients treated with apixaban and warfarin, respectively.
The incidence of ISTH major gastrointestinal bleeds (including upper GI, lower GI, and rectal bleeding) was lower with apixaban (0.76%/year) compared to warfarin (0.86%/year).
The incidence of ISTH major intraocular bleeding was higher with apixaban (0.18%/year) compared to warfarin (0.13%/year).
AVERROES Study: There was an increase in the incidence of major bleeding between the apixaban and ASA treatment group, which was not statistically significant (see Table 9). The frequency of fatal and intracranial bleeding was similar in the 2 treatment groups. (See Table 9.)
Click on icon to see table/diagram/image
Treatment discontinuation due to bleeding related adverse reactions occurred in 1.5% and 1.3% of patients treated with apixaban and ASA, respectively.
Subpopulation Analysis: In the ARISTOTLE study, the results for the primary efficacy endpoint and major bleeding results were generally consistent across all major subgroups including age, weight, CHADS
2 score, warfarin naive status, level of renal impairment, assignment to reduced dose apixaban, and ASA at randomization (see Figure 3).
Similarly, in the AVERROES study, the results for the primary efficacy endpoint and major bleeding results were consistent across all major subgroups including age, CHADS
2 score, level of renal impairment, and previous VKA use or VKA refusal (see Figure 4).
Notably, the efficacy and safety results for both studies in elderly patients (including those ≥75 years) were consistent with the overall population. (See Figure 3 and Figure 4.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Patients undergoing cardioversion: EMANATE, an open-label, multi-center study, enrolled 1500 patients who were either oralanticoagulant naïve or pre-treated less than 48 hours, and scheduled for cardioversion for NVAF.
Patients were randomized 1:1 to apixaban or to heparin and/or VKA for the prevention of cardiovascular events. Electrical and/or pharmacologic cardioversion was conducted after at least5 doses of 5 mg twice daily apixaban [or 2.5 mg twice daily in selected patients (see Dosage & Administration) or at least 2 hours after a 10 mg loading dose [or a 5 mg loading dose in selected patients (see Dosage & Administration)] if earlier cardioversion was required. In the apixaban group, 342 patients received a loading dose (331 patients received the 10 mg dose and 11 patients received the 5 mg dose).
There were no strokes (0%) in the apixaban group (n=753) and 6 (0.80%) strokes in the heparin and/or VKA group (n=747; RR 0, 95% CI 0.0, 0.64) (nominal p-value = 0.0151). All-cause death occurred in 2 patients (0.27%) in the apixaban group and 1 patient (0.13%) in the heparin and/or VKA group (RR 1.98, 95% CI 0.19, 54.00). No systemic embolism events were reported.
Major bleeding and CRNM bleeding events occurred in 3 (0.41%) and 11 (1.50%) patients,respectively, in the apixaban group, compared to 6 (0.83%) and 13 (1.80%) patients in the heparin and/or VKA group.
This exploratory study showed comparable efficacy and safety between apixaban and heparin and/or VKA treatment groups in the setting of cardioversion.
Treatment of VTE: The clinical program was designed to demonstrate the efficacy and safety of apixaban for the treatment of DVT and PE (AMPLIFY), and extended therapy for the prevention of recurrent DVT and PE following 6 to 12 months of anticoagulant treatment for DVT and/or PE (AMPLIFY-EXT). Both studies were randomized, parallel-group, double-blind multinational trials in patients with symptomatic proximal DVT and/or symptomatic PE. All key safety and efficacy endpoints were adjudicated by an independent blinded committee. (See Table 10 and Table 11.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
AMPLIFY Study: Patients were randomized to treatment with apixaban 10 mg twice daily orally for 7 days followed by apixaban 5 mg twice daily orally for 6 months, or enoxaparin 1 mg/kg twice daily subcutaneously for at least 5 days (until INR ≥2) and warfarin (target INR range 2.0-3.0) orally for 6 months. Patients who required thrombectomy, insertion of a caval filter, or use of a fibrinolytic agent, and patients with creatinine clearance <25 mL/min, significant liver disease, or active bleeding were excluded from the studies. Patients were allowed to enter the study with or without prior parenteral anticoagulation (up to 48 hours).
For patients randomized to warfarin, the mean percentage of time in therapeutic range (INR 2.0 3.0) was 60.9.
The primary objective of the study was to determine if apixaban was noninferior to enoxaparin/warfarin therapy in the combined endpoint of adjudicated recurrent symptomatic VTE (nonfatal DVT or nonfatal PE) or VTE-related death over 6 months of therapy.
In the study, apixaban was shown to be noninferior to enoxaparin/warfarin in the combined endpoint of adjudicated recurrent symptomatic VTE (nonfatal DVT or nonfatal PE) or VTE-related death (see Table 12).
Click on icon to see table/diagram/image
Figure 5 is a plot of the time from randomization to the occurrence of the first primary efficacy endpoint event in the two treatment groups in the AMPLIFY study. (See Figure 5.)
Click on icon to see table/diagram/image
Apixaban efficacy in initial treatment of VTE was consistent between patients who were treated for a PE [Relative Risk 0.9, 95% confidence interval (0.5, 1.6)] or DVT [Relative Risk 0.8, 95% confidence interval (0.5, 1.3)]. Efficacy across subgroups, including age, gender, renal function, body mass index (BMI), extent of index PE, location of DVT thrombus, and prior parenteral heparin use was generally consistent (see Figure 6).
Click on icon to see table/diagram/image
The primary safety endpoint was major bleeding. In the study, apixaban was statistically superior to enoxaparin/warfarin in the primary safety endpoint [Relative Risk 0.31, 95% confidence interval (0.17, 0.55), P-value <0.0001] (see Table 13).
Click on icon to see table/diagram/image
The adjudicated major bleeding and CRNM bleeding at any anatomical site was generally lower in the apixaban group compared to the enoxaparin/warfarin group. Adjudicated ISTH major gastrointestinal bleeding occurred in 6 (0.2%) apixaban-treated patients and 17 (0.6%) enoxaparin/warfarin-treated patients.
During the 6 months of the study, fewer patients were hospitalized in the apixaban group [153 (5.7%)] compared to the warfarin treated patients [190 (7.1%)].
AMPLIFY-EXT Study: Patients were randomized to treatment with apixaban 2.5 mg twice daily orally, apixaban 5 mg twice daily orally, or placebo for 12 months after completing 6 to 12 months of initial anticoagulant treatment. Approximately one-third of patients participated in the AMPLIFY study prior to enrollment in the AMPLIFY-EXT study.
The primary objective of the study was to determine if apixaban was superior to placebo in the combined endpoint of symptomatic, recurrent VTE (nonfatal DVT or nonfatal PE) or all-cause death.
In the study, both doses of apixaban were statistically superior to placebo in the primary endpoint of symptomatic, recurrent VTE or all-cause death (see Table 14).
Click on icon to see table/diagram/image
Figure 7 is a plot of the time from randomization to the occurrence of the first primary efficacy endpoint event in the three treatment groups in the AMPLIFY-EXT study. (See Figure 7.)
Click on icon to see table/diagram/image
Apixaban efficacy for prevention of a recurrence of a VTE was maintained across subgroups, including age, gender, BMI, and renal function.
The primary safety endpoint was major bleeding during the treatment period. In the study, the incidence in major bleeding was similar between the apixaban and placebo groups, there was no statistically significant difference in the incidence of major + CRNM, minor, and all bleeding between the apixaban 2.5 mg twice daily and placebo treatment groups. The frequency of major + CRNM bleeding in the apixaban 5 mg twice daily group was not statistically different from the placebo group. The frequency of CRNM, minor bleeding, and all bleeding in the apixaban 5 mg twice daily group was statistically different from the placebo group (see Table 15).
Click on icon to see table/diagram/image
Figure 8 is a plot of the time from randomization to the occurrence of the first major or clinically relevant non-major bleeding event in the three treatment groups in the AMPLIFY-EXT study. (See Figure 8.)
Click on icon to see table/diagram/image
ISTH major gastrointestinal bleeding occurred in 1 (0.1%) apixaban-treated patient at the 5 mg twice daily dose, no patients at the 2.5 mg twice daily dose, and 1 (0.1%) placebo-treated patient.
During the 12 months of the study, fewer patients were hospitalized in the apixaban groups [42 (5%) in the 2.5 mg twice daily group; 34 (4.2%) in the 5 mg twice daily group] compared to the placebo treated patients [62 (7.5%)].
Pharmacokinetics: Absorption: The absolute bioavailability of apixaban is approximately 50% for doses up to 10 mg. Apixaban is rapidly absorbed with maximum concentrations (C
max) appearing 3 to 4 hours after tablet intake. Intake with food does not affect apixaban AUC or C
max at the 10 mg dose. Apixaban can be taken with or without food. Apixaban demonstrates linear pharmacokinetics with dose proportional increases in exposure for oral doses up to 10 mg. At doses ≥25 mg, apixaban displays dissolution limited absorption with decreased bioavailability. Apixaban exposure parameters exhibit low to moderate variability reflected by a within-subject and inter-subject variability of ~20% CV and ~30% CV, respectively.
Following oral administration of 10 mg of apixaban as 2 crushed 5 mg tablets suspended in 30 mL of water, exposure was comparable to exposure after oral administration of 2 intact 5 mg tablets.
Following oral administration of 10 mg of apixaban as 2 crushed 5 mg tablets mixed with 30 g of apple sauce, the C
max and AUC were 21% and 16% lower, respectively, when compared to administration of 2 intact 5 mg tablets.
Following administration of a crushed 5 mg apixaban tablet suspended in 60 mL of D5W and delivered via a nasogastric tube, exposure was similar to exposure seen in other clinical trialsinvolving healthy subjects receiving a single oral 5 mg apixaban tablet dose.
Distribution: Plasma protein binding in humans is approximately 87%. The volume of distribution (Vss) is approximately 21 liters.
Metabolism and Elimination: Apixaban has multiple routes of elimination. Of the administered apixaban dose in humans, approximately 25% was recovered as metabolites, with the majority recovered in feces. Renal excretion of apixaban accounts for approximately 27% of total clearance. Additional contributions from biliary and direct intestinal excretion were observed in clinical and nonclinical studies, respectively.
Apixaban has a total clearance of about 3.3 L/h and a half-life of approximately 12 hours.
O-demethylation and hydroxylation at the 3-oxopiperidinyl moiety are the major sites of biotransformation. Apixaban is metabolized mainly via CYP3A4/5 with minor contributions from CYP1A2, 2C8, 2C9, 2C19, and 2J2. Unchanged apixaban is the major drug-related component in human plasma with no active circulating metabolites present. Apixaban is a substrate of transport proteins, P-gp and breast cancer resistance protein (BCRP).
Renal Impairment: There was no impact of impaired renal function on peak concentration of apixaban. There was an increase in apixaban exposure correlated to decrease in renal function, as assessed via measured creatinine clearance. In individuals with mild (creatinine clearance 51-80 mL/min), moderate (creatinine clearance 30-50 mL/min) and severe (creatinine clearance 15-29 mL/min) renal impairment, apixaban plasma concentrations (AUC) were increased 16, 29, and 44%, respectively, compared to individuals with normal creatinine clearance. Renal impairment had no evident effect on the relationship between apixaban plasma concentration and anti-FXa activity. No dose adjustment is necessary in patients with mild, moderate or severe renal impairment, except as described in Dosage & Administration.
In subjects with end-stage renal disease (ESRD), the AUC of apixaban was increased by 36% when a single dose of apixaban 5 mg was administered immediately after hemodialysis, compared to that seen in subjects with normal renal function. Hemodialysis, started two hours after administration of a single dose of apixaban 5 mg, decreased apixaban AUC by 14% in these ESRD subjects, corresponding to an apixaban dialysis clearance of 18 mL/min.
Hepatic Impairment: Apixaban has not been studied in patients with severe hepatic impairment or active hepatobiliary disease. Apixaban is not recommended in patients with severe hepatic impairment (see Precautions).
In a study comparing subjects with mild and moderate hepatic impairment (classified as Child Pugh A and B, respectively) to healthy control subjects, the single-dose pharmacokinetics and pharmacodynamics of apixaban 5 mg were not altered in subjects with hepatic impairment. Changes in anti-FXa activity and INR were comparable between subjects with mild to moderate hepatic impairment and healthy subjects. No dose adjustment is required in patients with mild or moderate hepatic impairment; however, given the limited number of subjects studied, caution is advised when using ELIQUIS in this population (see Dosage & Administration and Precautions).
Elderly: Elderly patients (above 65 years) exhibited higher plasma concentrations than younger patients, with mean AUC values being approximately 32% higher. No dose adjustment is required, except as described in Dosage & Administration.
Gender: Exposure to apixaban was approximately 18% higher in females than in males. No dose adjustment is required.
Ethnic Origin and Race: The results across phase 1 studies showed no discernible difference in apixaban pharmacokinetics between White/Caucasian, Asian and Black/African American subjects. Findings from a population pharmacokinetic analysis in patients who received apixaban were generally consistent with the phase 1 results. No dose adjustment is required.
Body Weight: Compared to apixaban exposure in subjects with body weight of 65 to 85 kg, body weight >120 kg was associated with approximately 30% lower exposure and body weight <50 kg was associated with approximately 30% higher exposure. No dose adjustment is required, except as described in Dosage & Administration.
Pharmacokinetic/Pharmacodynamic Relationship: The pharmacokinetic/pharmacodynamic (PK/PD) relationship between apixaban plasma concentration and several PD endpoints (anti-FXa activity, INR, PT, aPTT) has been evaluated after administration of a wide range of doses (0.5-50 mg). The relationship between apixaban plasma concentration and anti-FXa activity was best described by a linear model. The PK/PD relationship observed in patients who received apixaban in Phase 2 or Phase 3 clinical trials was consistent with that established in healthy subjects.
Toxicology: Non-Clinical Safety: Preclinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, fertility and embryo fetal development (see Use in Pregnancy & Lactation). In the offspring of pregnant rats treated with apixaban there were decreases in mating and fertility. These effects were minimal and observed only at exposures considered sufficiently in excess of the maximum human exposure indicating little relevance to clinical use.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image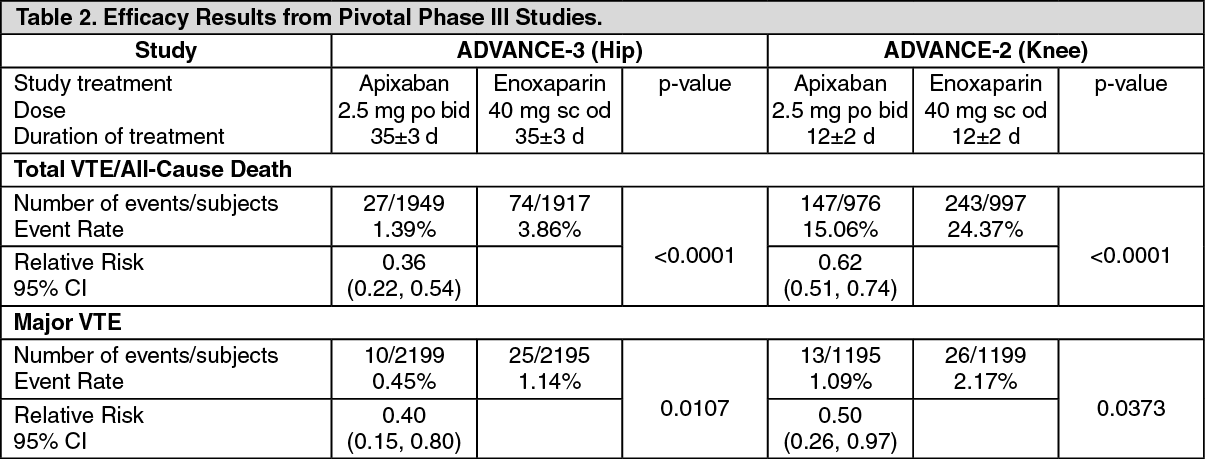 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image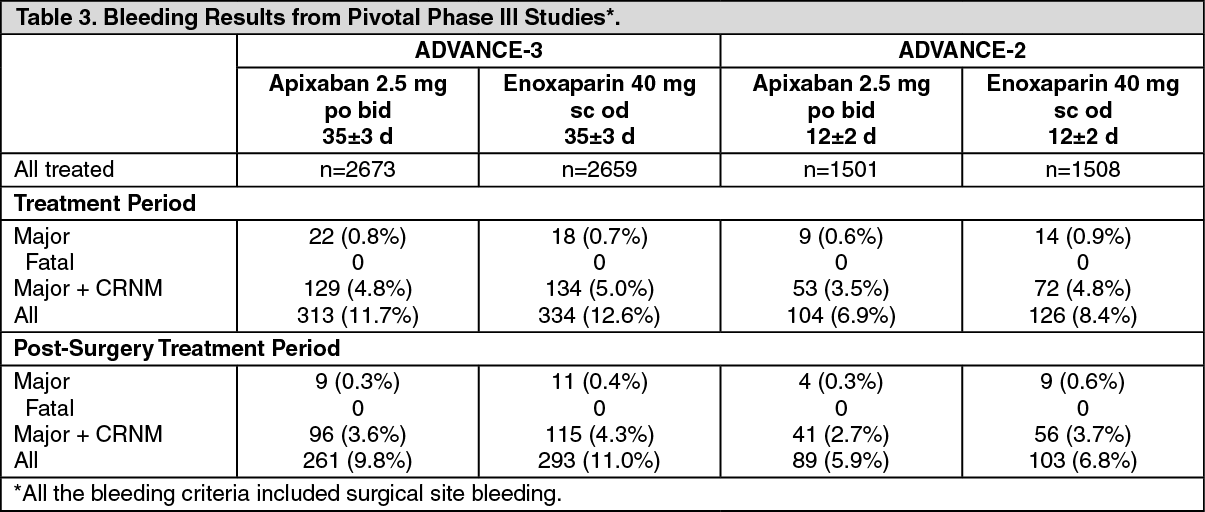 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image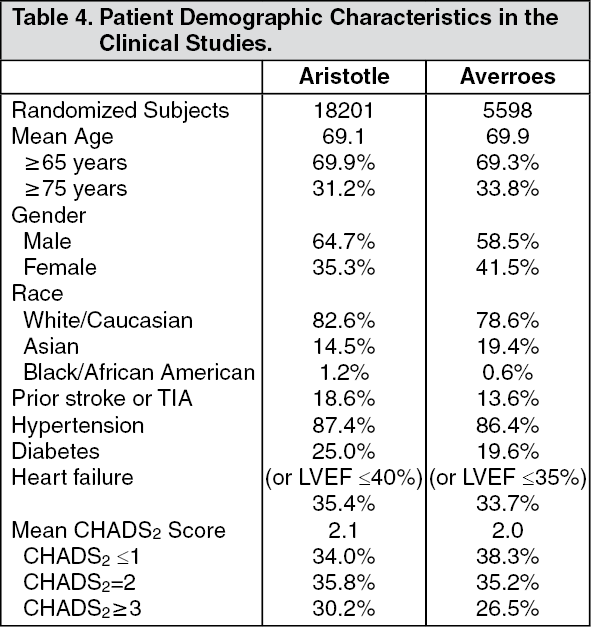 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image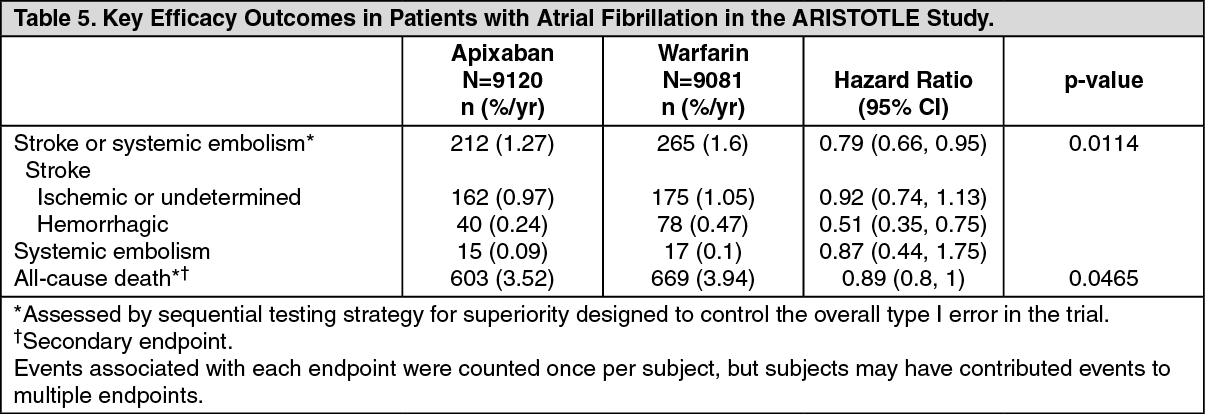 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image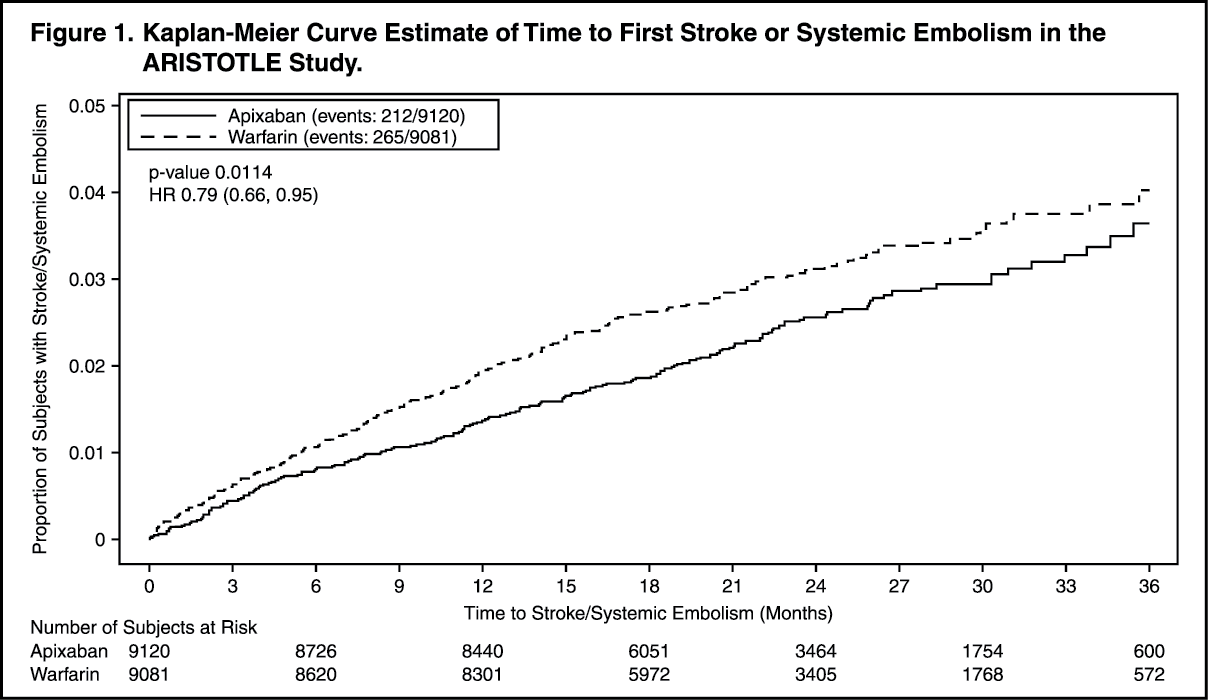 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image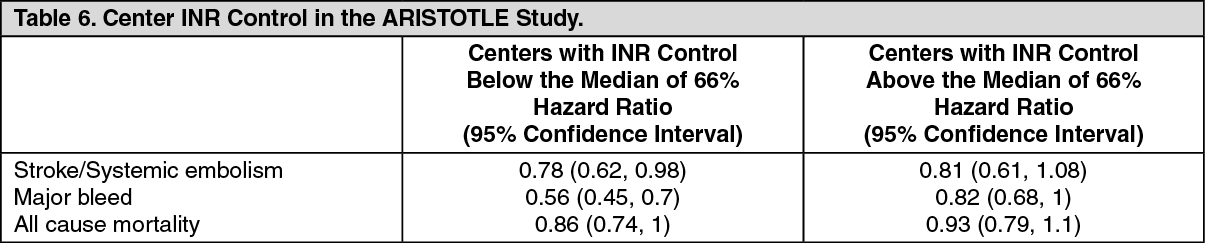 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image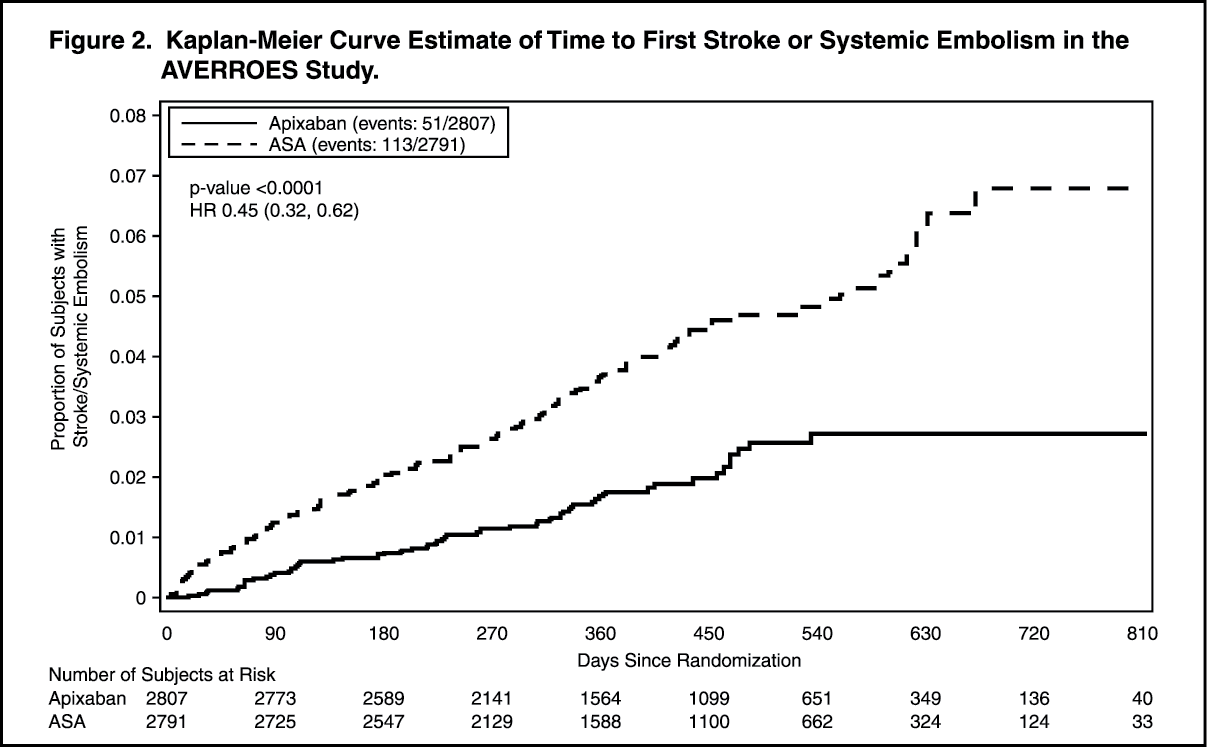 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image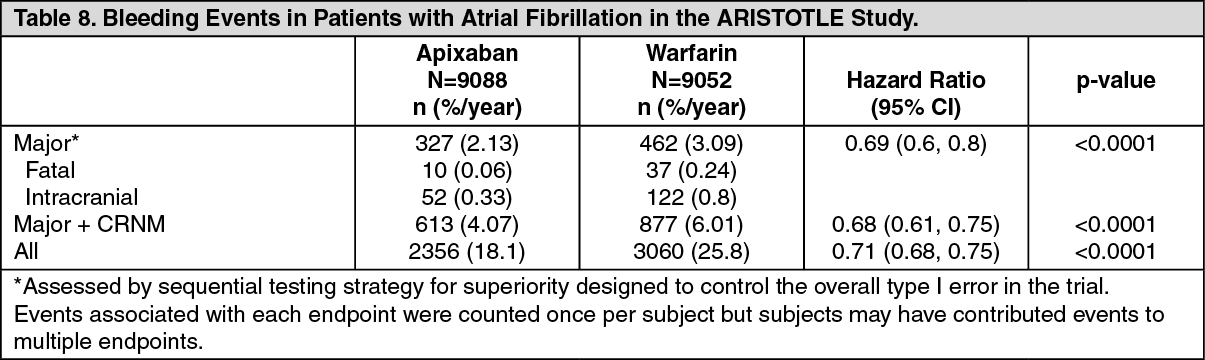 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image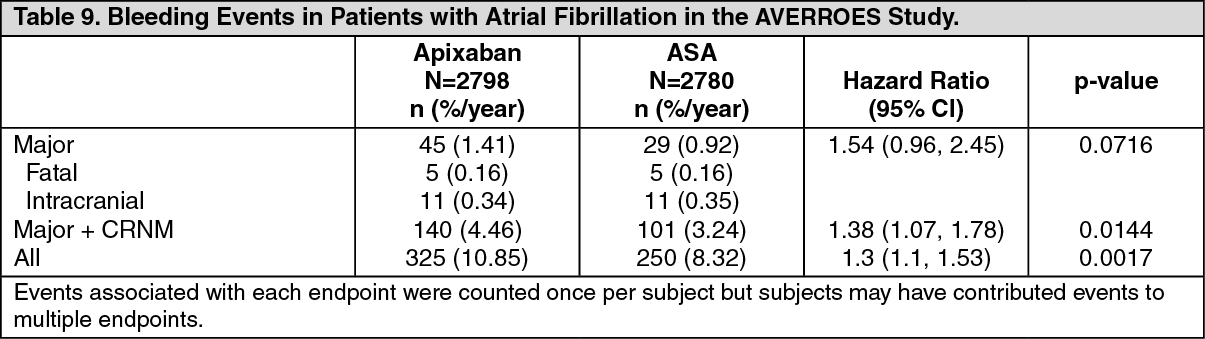 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image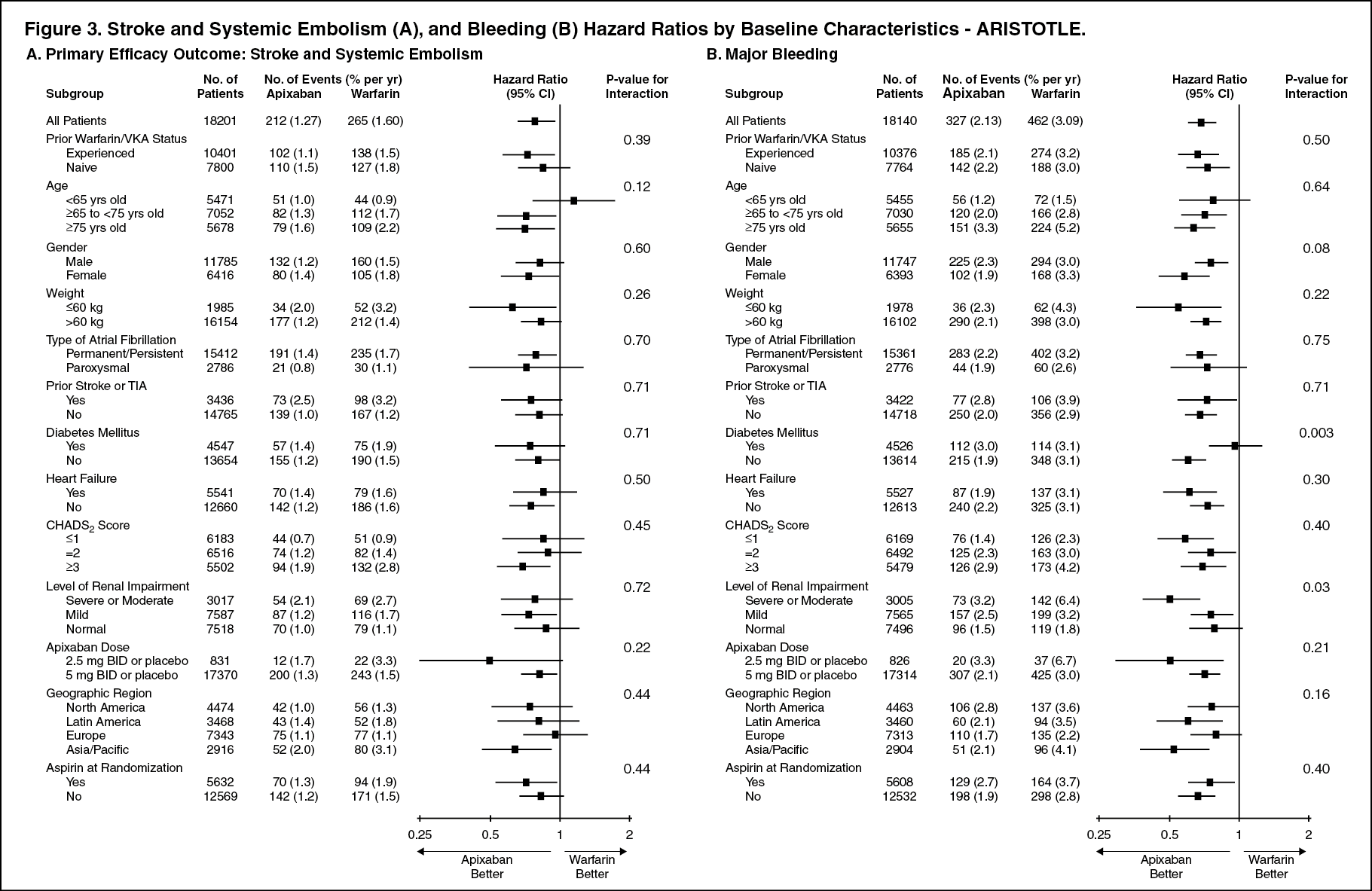 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image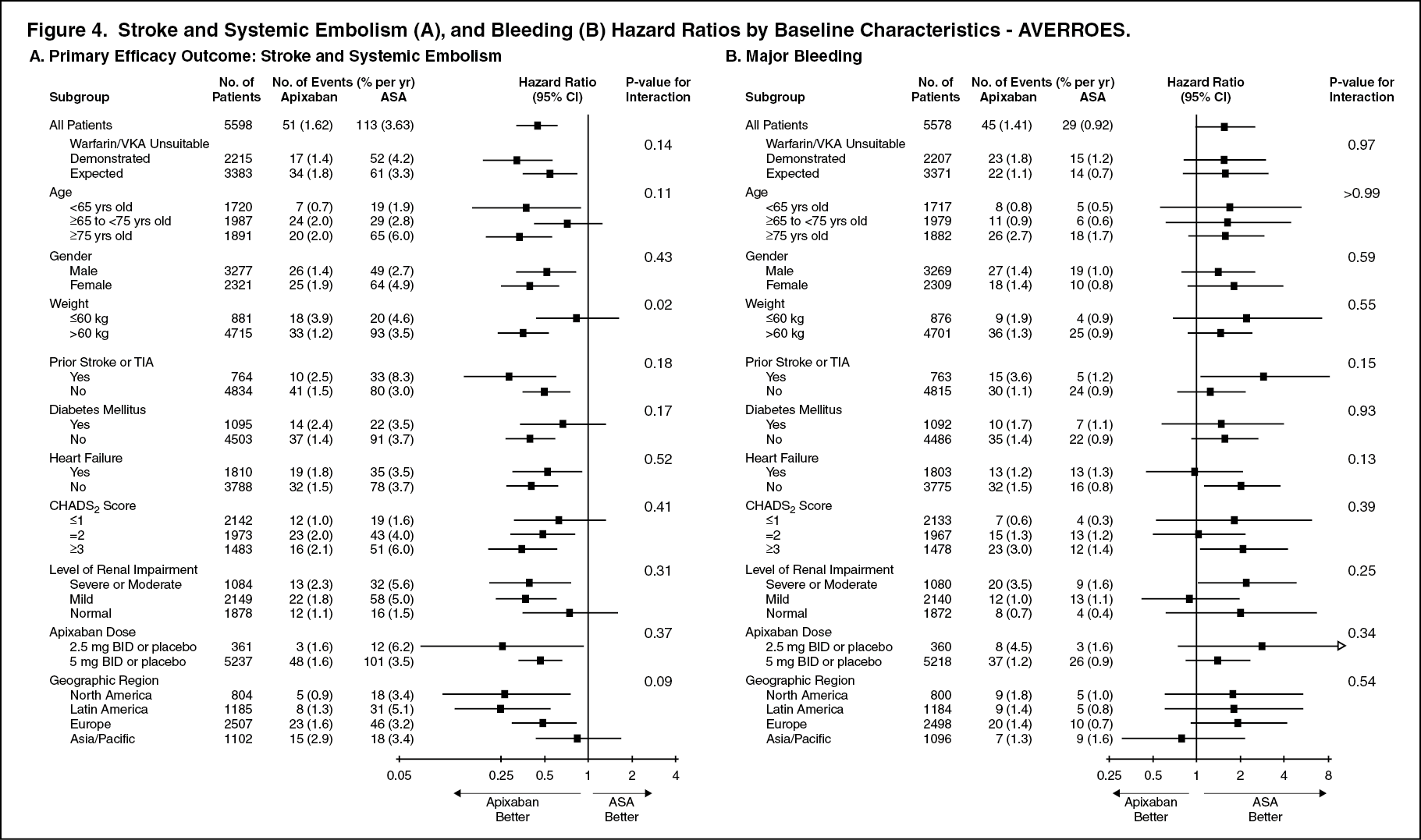 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image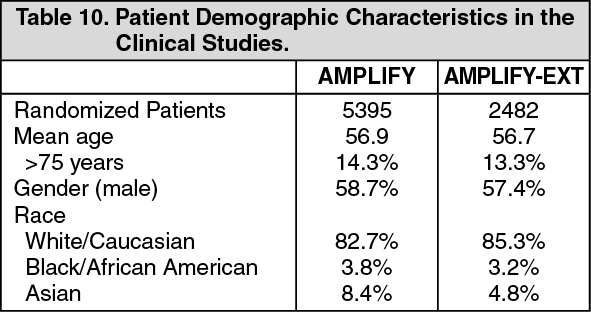 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image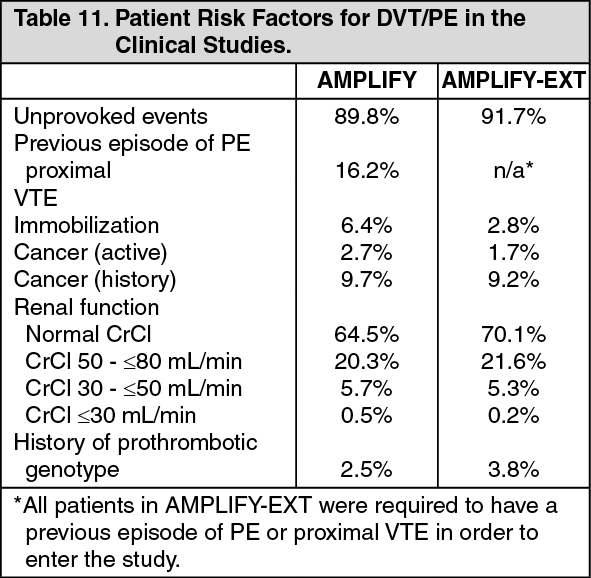 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image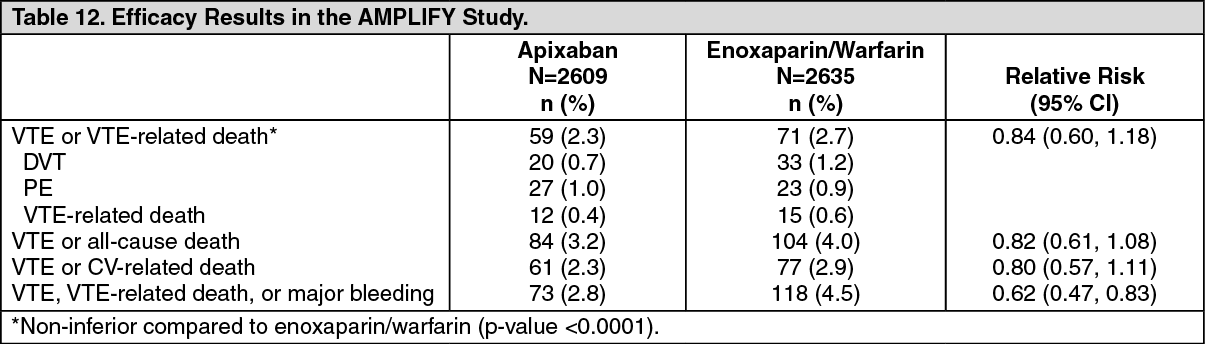 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image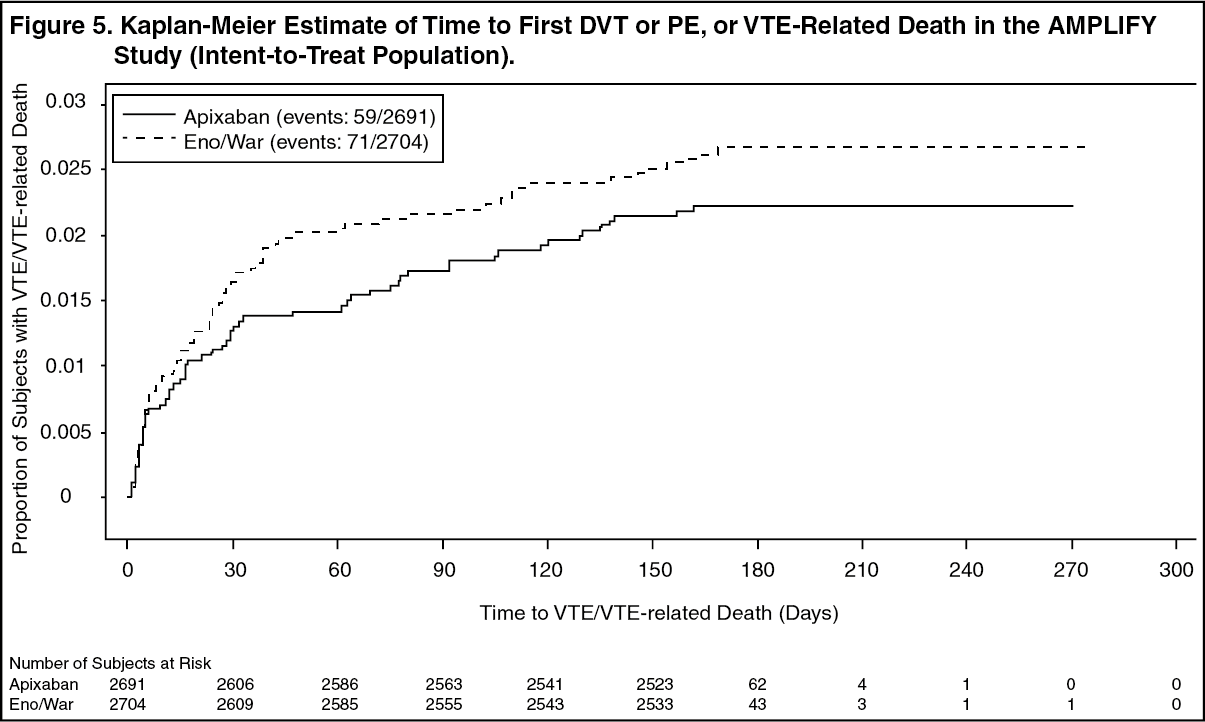 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image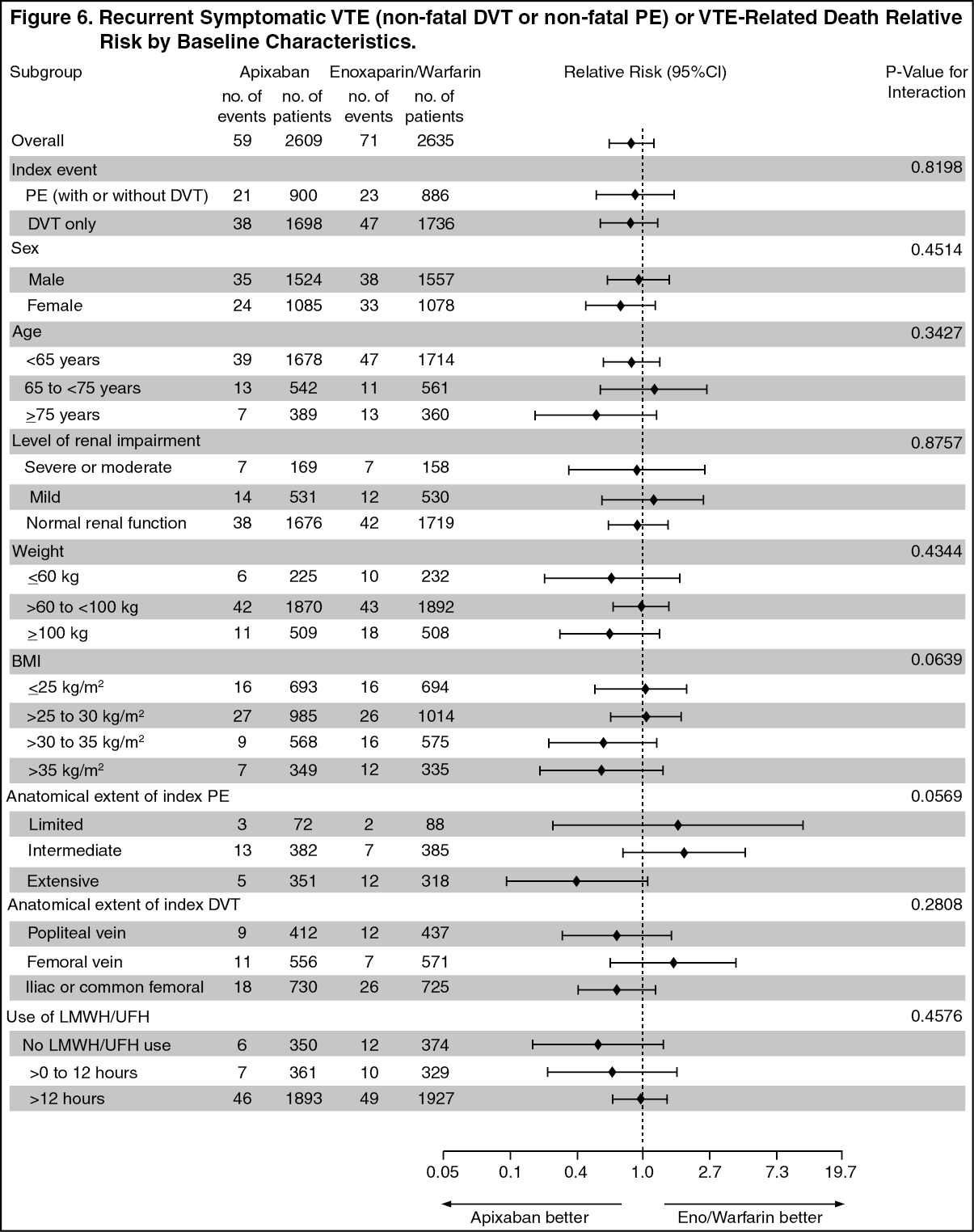 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image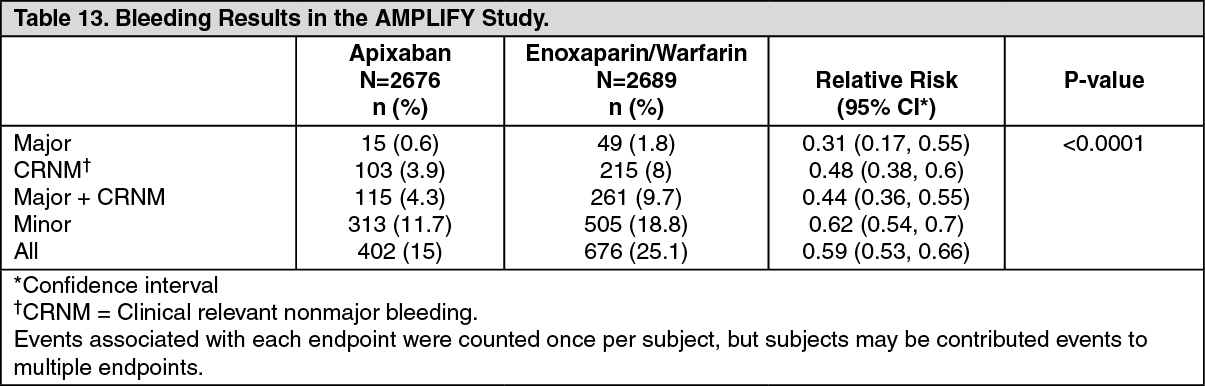 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image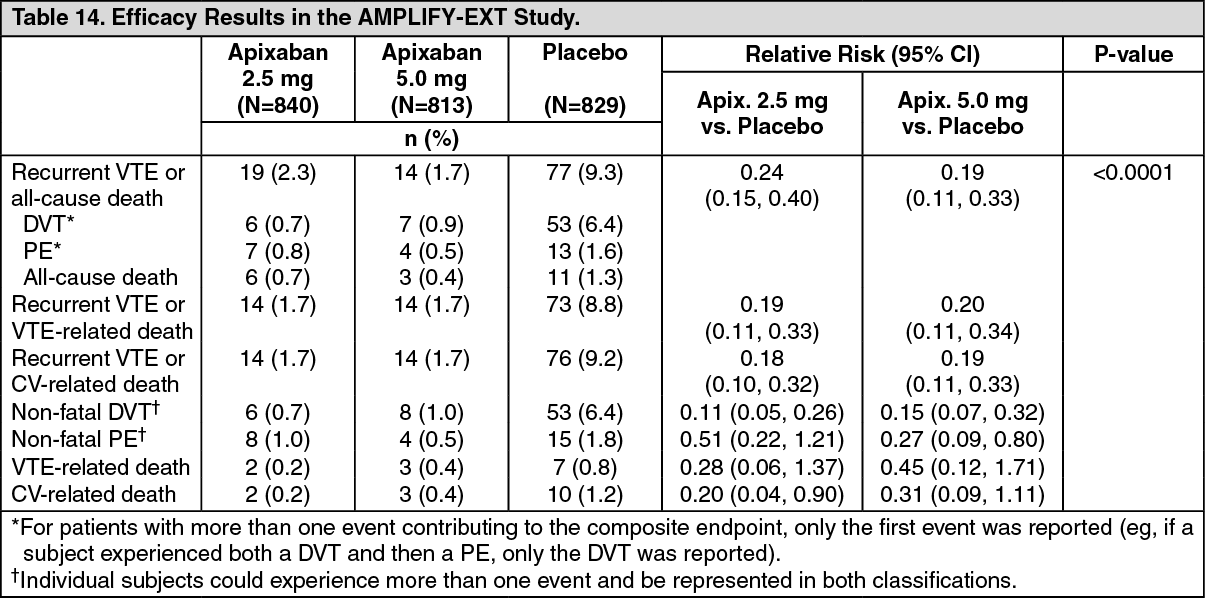 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image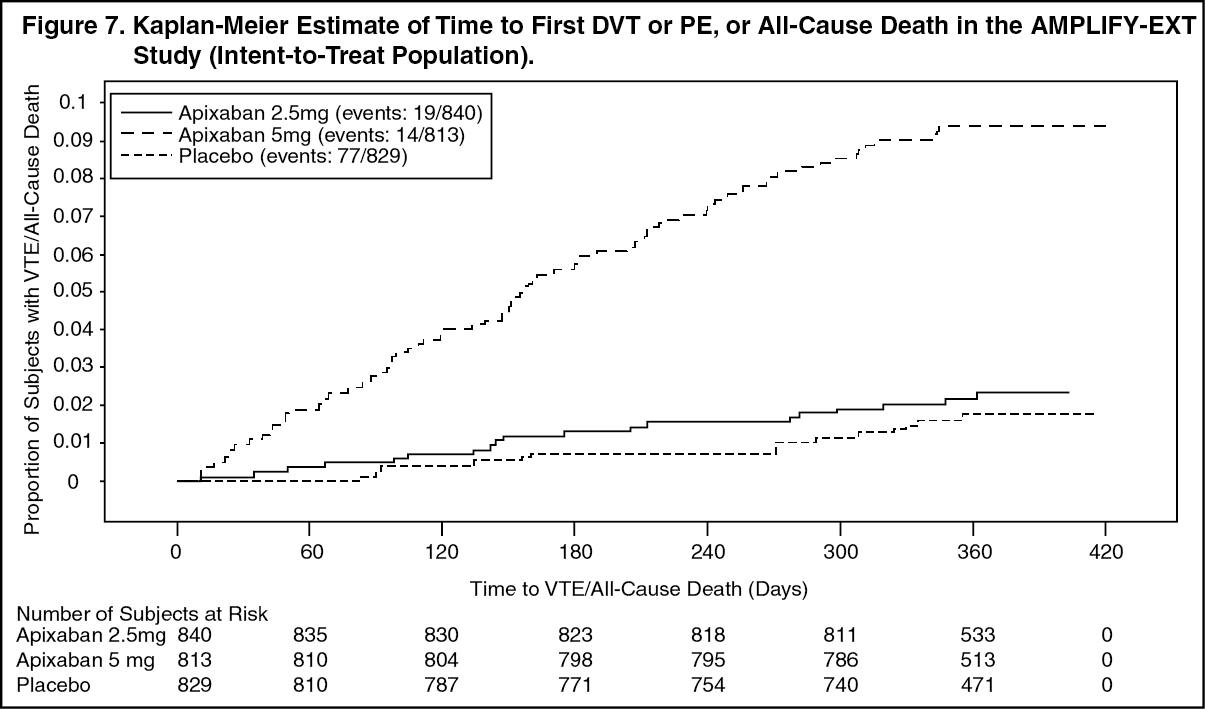 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image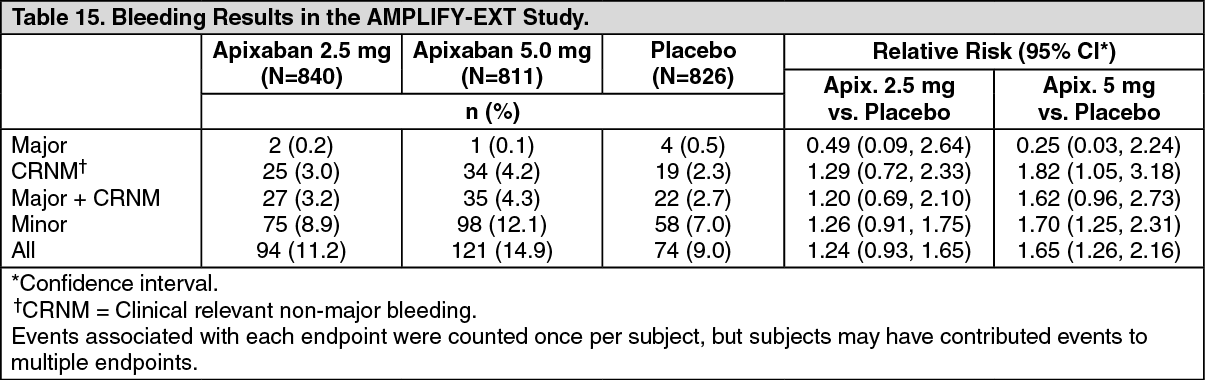 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image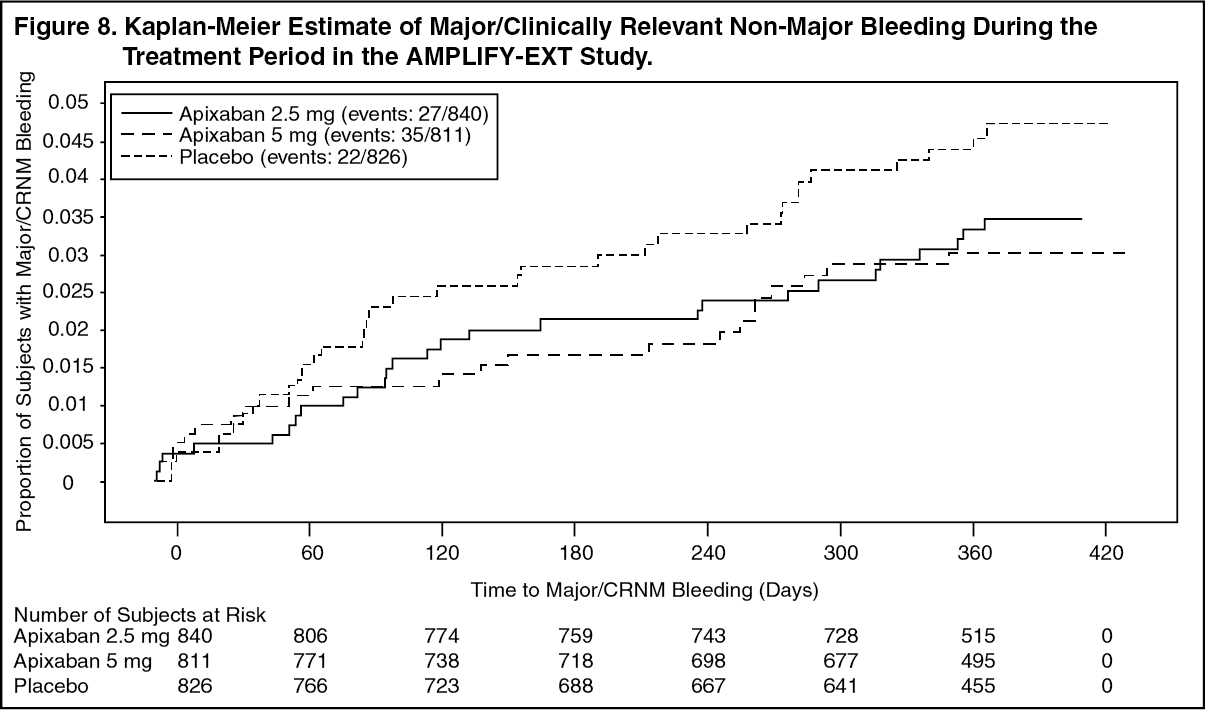 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image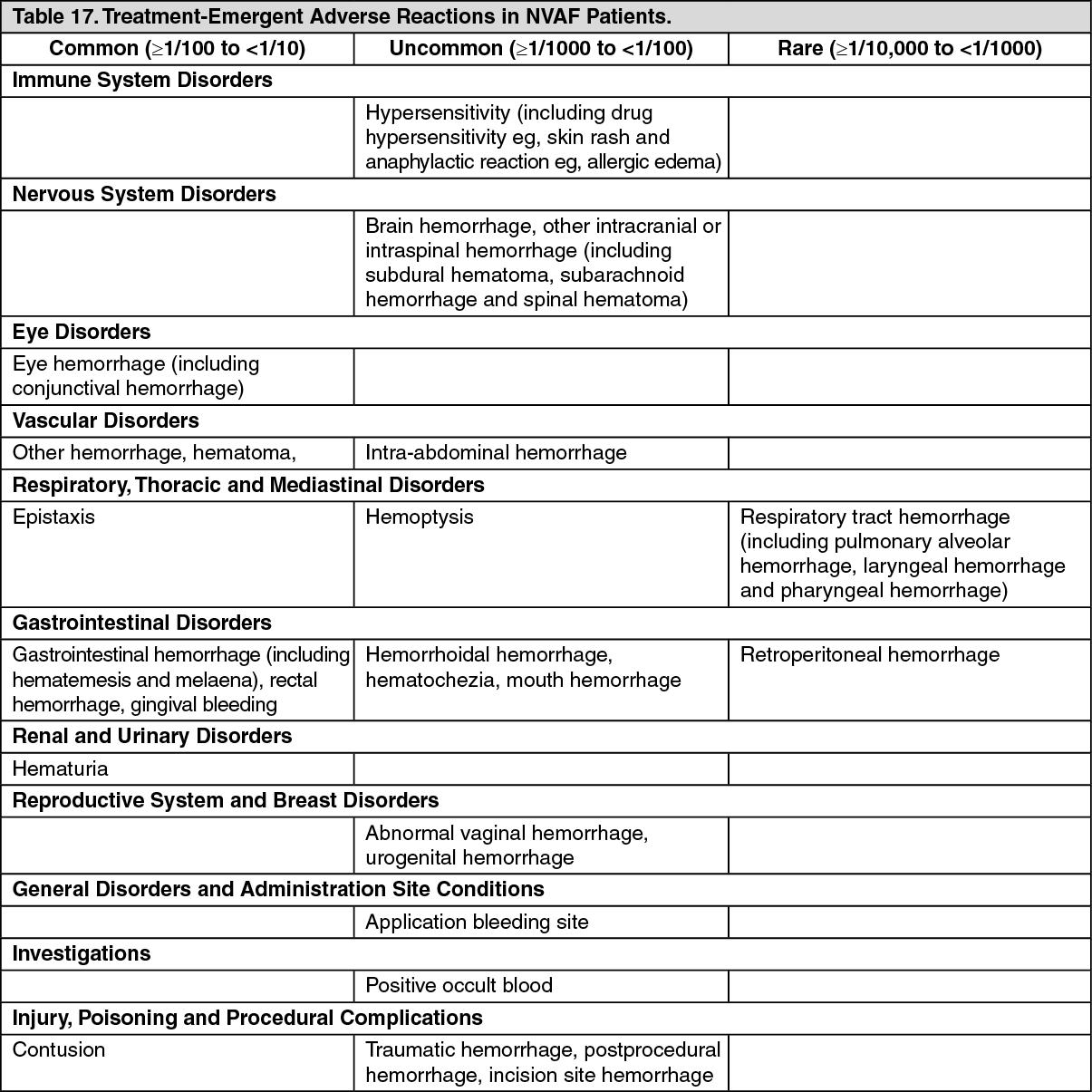 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image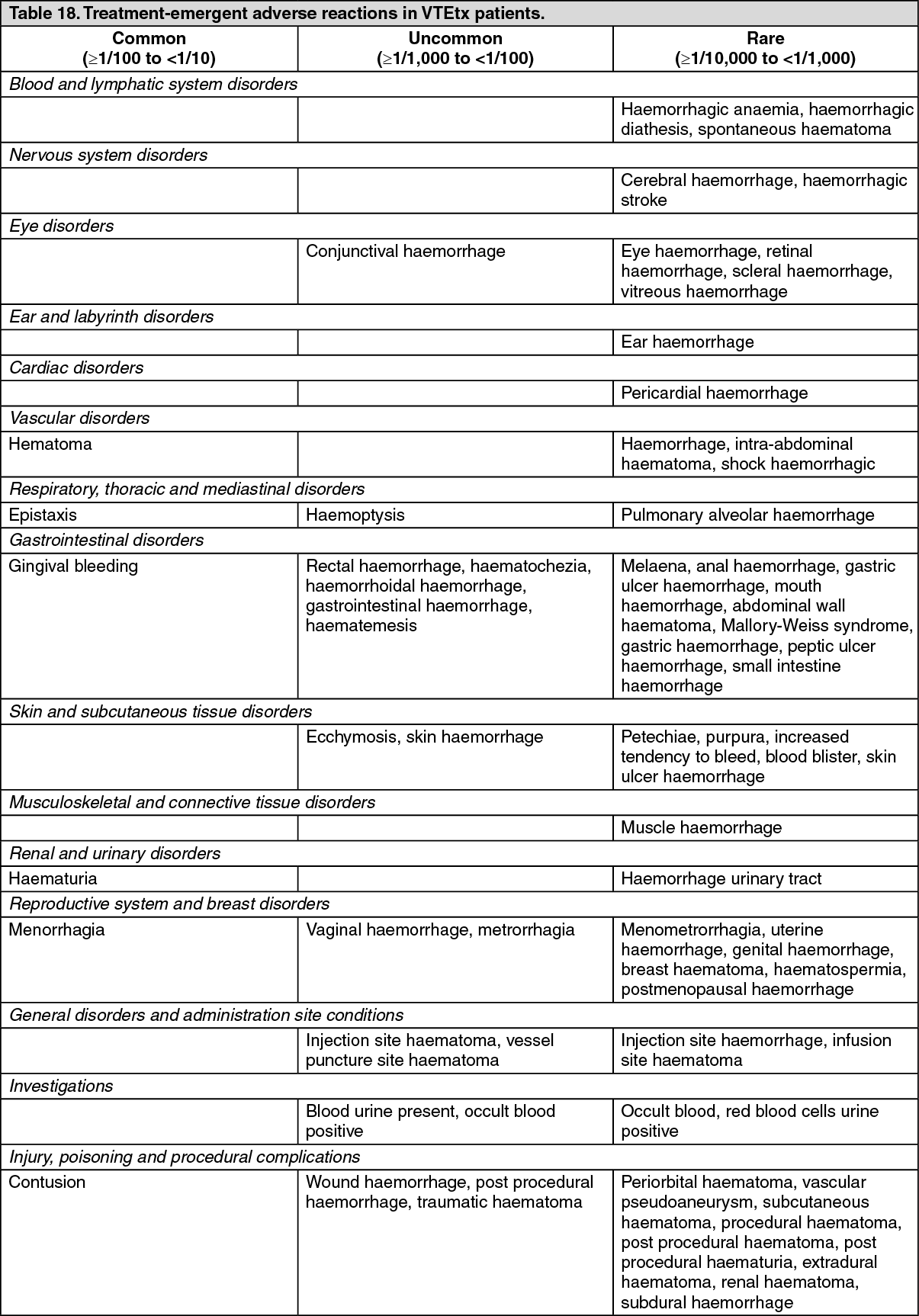 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out
