


 Sign Out
Sign Out
Mechanism of Action: Satralizumab is a humanized IgG2 monoclonal antibody (mAb) that binds to soluble and membrane-bound human IL-6 receptor (IL-6R), and thereby prevents IL-6 downstream signaling through these receptors.
IL-6 is a pleiotropic cytokine produced by a variety of cell types and is involved in diverse inflammatory processes including B-cell activation, differentiation of B-cells to plasmablasts and production of autoantibodies, Th17-cell activation and differentiation, T-regulatory cell inhibition, and changes in blood-brain-barrier permeability. IL-6 levels are increased in cerebrospinal fluid and serum of patients with NMO and NMOSD during periods of disease activity. Some IL-6 functions have been implicated in the pathogenesis of NMO and NMOSD, including production of pathological autoantibodies against Aquaporin-4 (AQP4), a water channel protein mainly expressed by astrocytes in the CNS.
Clinical/Efficacy Studies: The efficacy and safety of Enspryng were evaluated in two pivotal phase III clinical trials (BN40898 and BN40900) in patients with a diagnosis of AQP4-IgG seropositive or seronegative NMO (Wingerchuck 2006 criteria), or with a diagnosis of AQP4-IgG seropositive NMOSD (Wingerchuk 2007 criteria). In retrospect, these patients also met the latest criteria proposed by the international panel for NMO diagnosis (Wingerchuk et al 2015). The effect of Enspryng was studied in adult (studies BN40898 and BN40900) and adolescent (aged ≥12 to <18 years) patients (study BN40898). The inclusion of AQP4-IgG seronegative adult NMO patients was limited to approximately 30% in both studies in order for the study population to reflect the real-world NMO patient population.
The primary efficacy measure in both studies was protocol-defined relapses (PDR) based on a pre-specified worsening in the Expanded Disability Status Scale (EDSS) and Functional System Scores (FSS) and confirmed by an independent Clinical Endpoint Committee (CEC). The primary endpoint analysis was time to first CEC-confirmed PDR with EDSS/FSS assessment performed within 7 days after symptoms were reported by the patient (adjudicated relapse).
Study BN40898 (also known as SA-307JG or SAkuraSky): Study BN40898 was a randomized, multicenter, double-blind, placebo-controlled clinical trial to evaluate the effect of Enspryng in combination with stable IST (OCs up to 15 mg/day [prednisolone equivalent], AZA up to 3 mg/kg/day or MMF up to 3000 mg/day; adolescents received a combination of AZA and OCs or MMF and OCs). The study included 83 AQP4-IgG seropositive and seronegative patients (including 7 adolescents). Patients received the first 3 single doses of Enspryng 120 mg or matching placebo by SC injection in the abdominal or femoral region every 2 weeks for the first 4 weeks and once every 4 weeks thereafter.
Study design and baseline characteristics of the study population are presented in Table 1.
The study was event-driven and the double-blind study period for efficacy evaluation ended when a total of 26 adjudicated relapses were observed. Patients who experienced a CEC-confirmed PDR or received rescue therapy for a relapse during the double-blind (DB) period or completed the DB period could enter the open-label extension period (OLE) where all patients received open-label treatment with Enspryng. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageStudy BN40900 (also known as SA-309JG or SAkuraStar): Study BN40900 was a randomized, multicenter, double-blind, placebo-controlled clinical trial to evaluate the effect of Enspryng monotherapy compared to placebo. The study included 95 AQP4-IgG seropositive and seronegative adult patients. Patients received the first 3 single doses of Enspryng 120 mg or matching placebo by SC injection in the abdominal or femoral region every 2 weeks for the first 4 weeks and once every 4 weeks thereafter.
Study design and baseline characteristics of the study population are presented in Table 2.
The double-blind study period for efficacy evaluation ended 1.5 years after the date of randomization of the last enrolled patient. Patients who experienced a CEC-confirmed PDR during the DB period or completed the DB period could enter the OLE period where all patients received open-label treatment with Enspryng. (See Table 2.)
Click on icon to see table/diagram/imagePrimary Efficacy – Double-Blind Period: Treatment with Enspryng resulted in a statistically significant 62% reduction in the risk of experiencing an adjudicated relapse (Hazard ratio [HR] [95% CI]: 0.38 [0.16-0.88]; p [log rank]=0.0184) when administered in combination with stable IST (Study BN40898) and 55% reduction in the risk of adjudicated relapse (HR [95% CI]: 0.45 [0.23-0.89]; p [log rank]=0.0184) when used as monotherapy (Study BN40900) when compared to placebo. At 48 weeks, 88.9% and 76.1% of Enspryng-treated patients remained adjudicated relapse-free when used in combination with IST or as monotherapy, respectively. At 96 weeks 77.6% and 72.1% of Enspryng-treated patients remained adjudicated relapse-free when used in combination with IST or as monotherapy, respectively. When data from the two studies were pooled, Enspryng treatment resulted in a 58% reduction in risk of adjudicated relapse compared to placebo (HR [95% CI]: 0.42 [0.25-0.71]; p [log rank]=0.0008) (see Table 3, Figure 1, Figure 2).
The strongest subgroup effect was observed in AQP4-IgG seropositive patients. In AQP4- IgG seropositive patients the relative risk of experiencing an adjudicated relapse in Study BN40898 was reduced by 79% (HR [95% CI]: 0.21 [0.06-0.75]), in Study BN40900 by 74% (HR [95% CI]: 0.26 [0.11-0.63]). At 48 weeks, 91.5% and 82.9% of Enspryng-treated AQP4-IgG seropositive patients remained adjudicated relapse-free when used in combination with IST or as monotherapy, respectively. At 96 weeks 91.5% and 76.5% of Enspryng-treated AQP4-IgG seropositive patients remained adjudicated relapse-free when used in combination with IST or as monotherapy, respectively. When data across studies BN40898 and BN40900 were pooled, treatment with Enspryng with or without IST led to an overall risk reduction of 75% (HR [95% CI]; 0.25 [0.12-0.50]) in AQP4-IgG seropositive patients (see Table 3, Figure 3, Figure 4). Differences in the time to first adjudicated relapse in AQP4-IgG seronegative patients between those patients receiving Enspryng with or without IST and those receiving placebo with or without IST were not significant (BN40898 and BN40900 pooled: HR [95% CI]: 0.97 [0.41-2.33]). (See Table 3, Figures 1, 2, 3 and 4.)
Click on icon to see table/diagram/image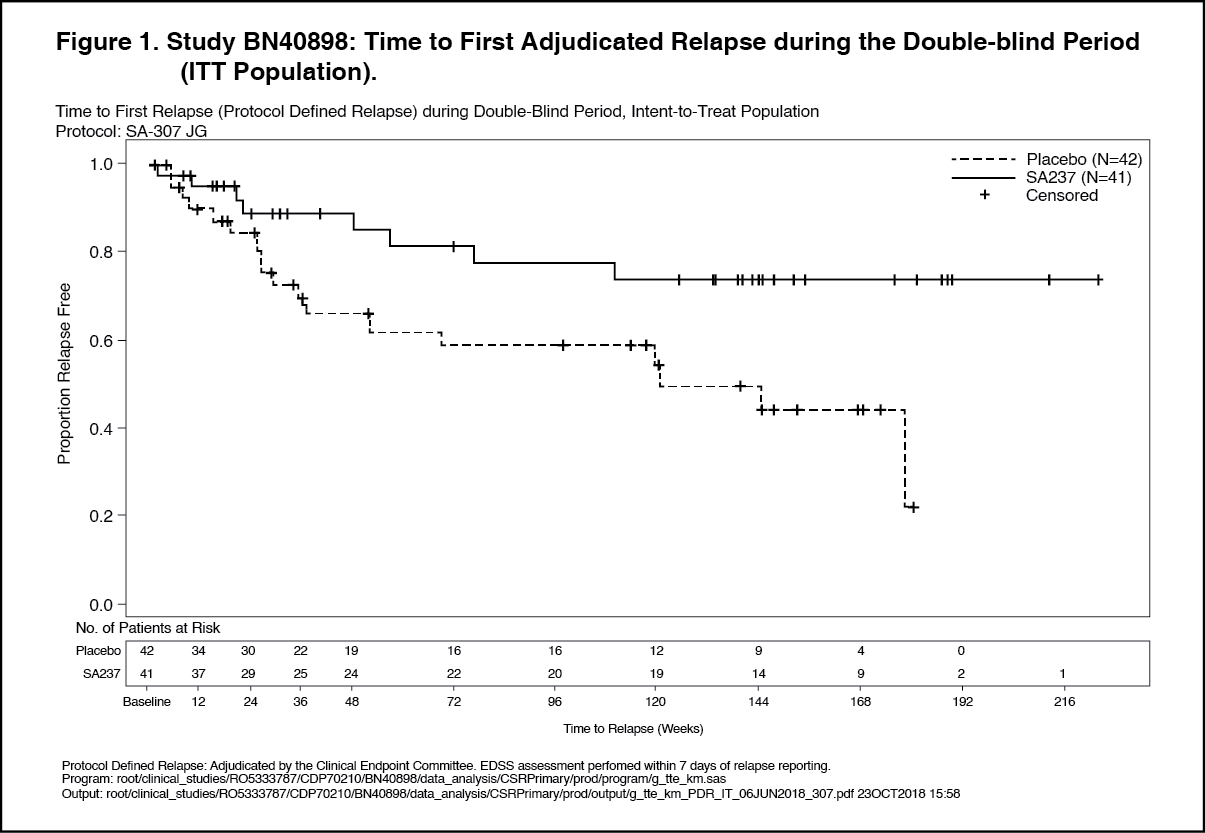 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image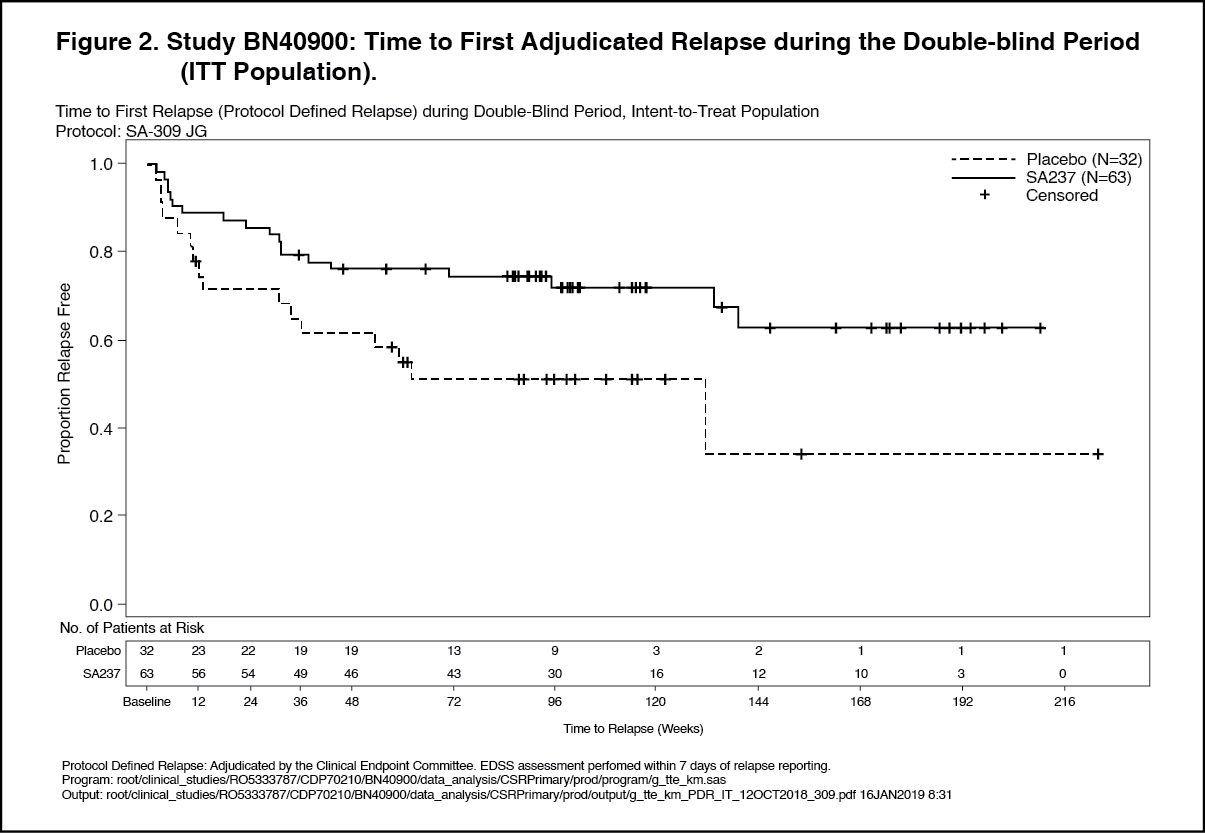 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image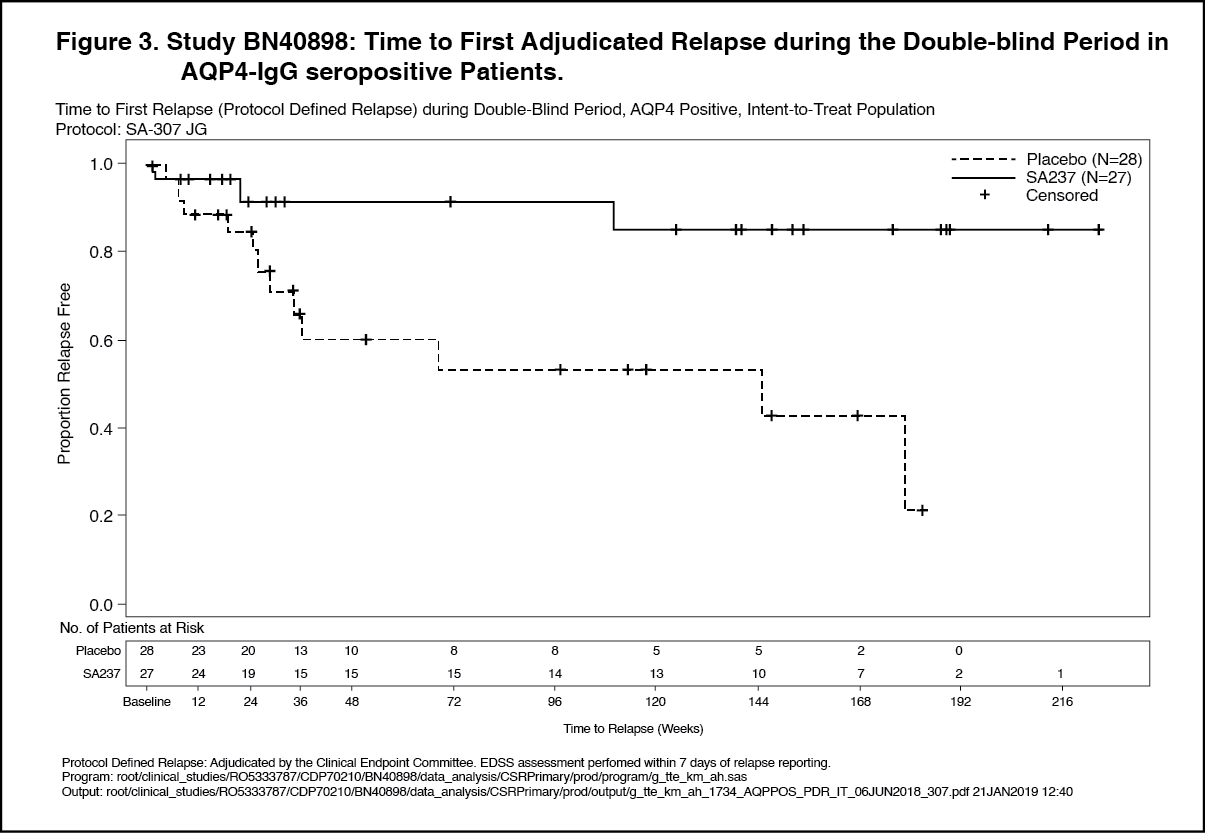 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image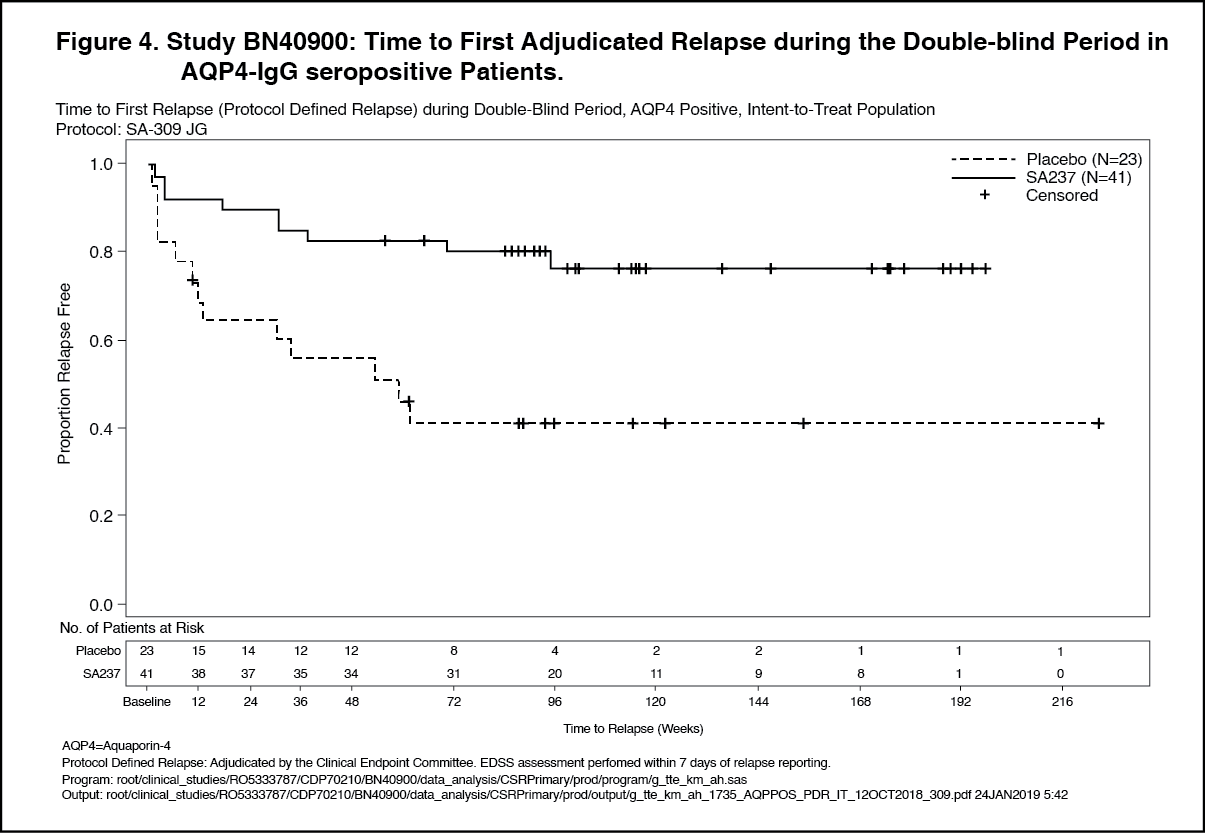 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageTreatment with Enspryng reduced the annualized rate of adjudicated relapses (ARR) by 74% in Study BN40898 and 73% in Study BN40900 compared to treatment with placebo (Table 4). The relative reduction in ARR in the AQP4-IgG seropositive subgroup was 88% and 90% in Studies BN40898 and BN40900 respectively. (See Table 4.)
Click on icon to see table/diagram/imageAs compared to placebo-treated patients, the need for rescue therapy (e.g., corticosteroids, intravenous immunoglobulin, and/or apheresis [including plasmapheresis or plasma exchange]) was reduced in Enspryng-treated patients by 51% in Study BN40898 and by 55% in Study BN40900 (ITT population). In the AQP4-IgG seropositive subgroup, ENSPRYNG treatment reduced the need for rescue therapy by 61% and 74% in Studies BN40898 and BN40900 respectively (Table 5). (See Table 5.)
Click on icon to see table/diagram/imageTreatment with Enspryng reduced the risk of experiencing a severe relapse defined as an EDSS increase ≥ 2 points from the previous EDSS assessment by 84% in study BN40898 and by 74% in study BN40900 compared to treatment with placebo (Table 6). The relative reduction in severe relapses in AQP4-IgG seropositive patients was 85% and 79% in studies BN40898 and BN40900, respectively. (See Table 6.)
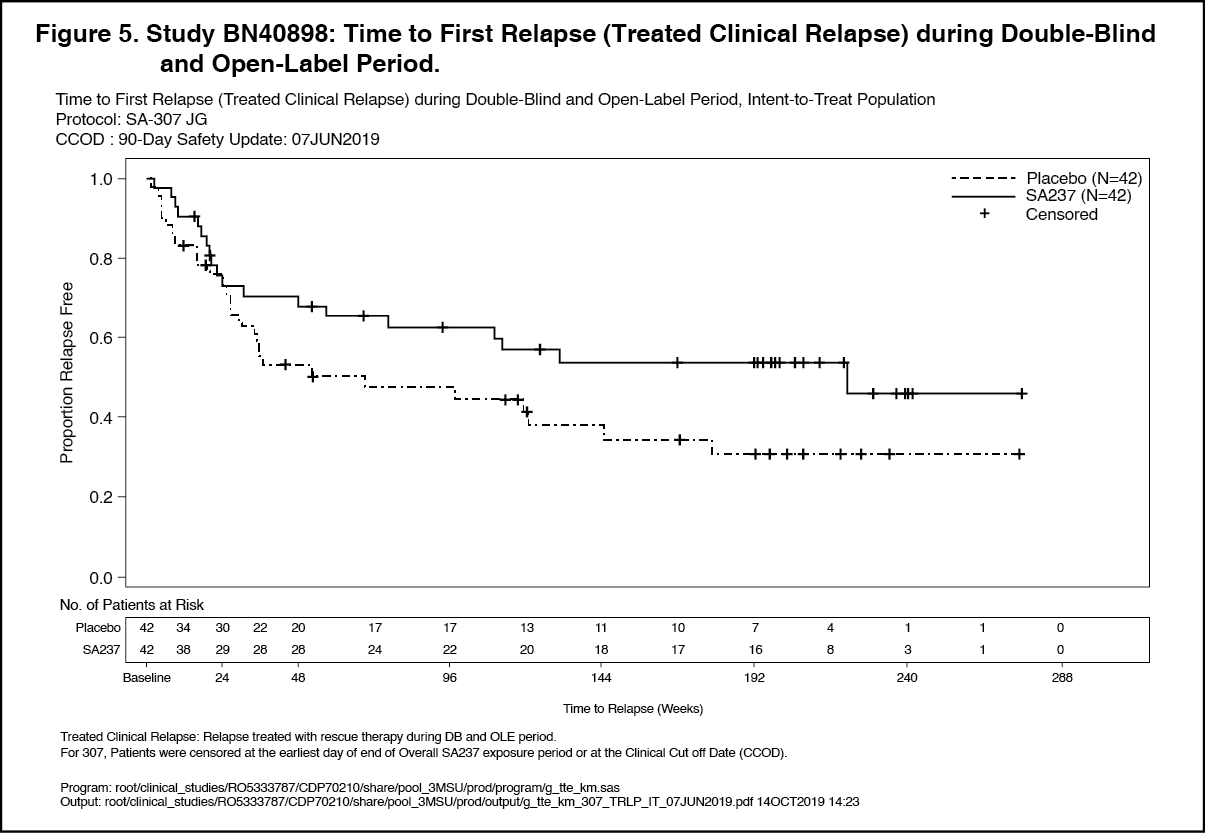
Click on icon to see table/diagram/imageOpen-Label Extension: Analyses of longer term data including the OLE period (based on relapse treated with rescue therapy) showed that 57% and 71% of patients treated with Enspryng remained relapse-free after 120 weeks of treatment, when Enspryng was administered as add-on therapy or as monotherapy, respectively.
In the AQP4-IgG seropositive population, 58% and 73% of patients remained relapse free after 120 weeks of treatment with Enspryng administered as add-on therapy or as monotherapy, respectively. (See Figures 5, 6, 7 and 8.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image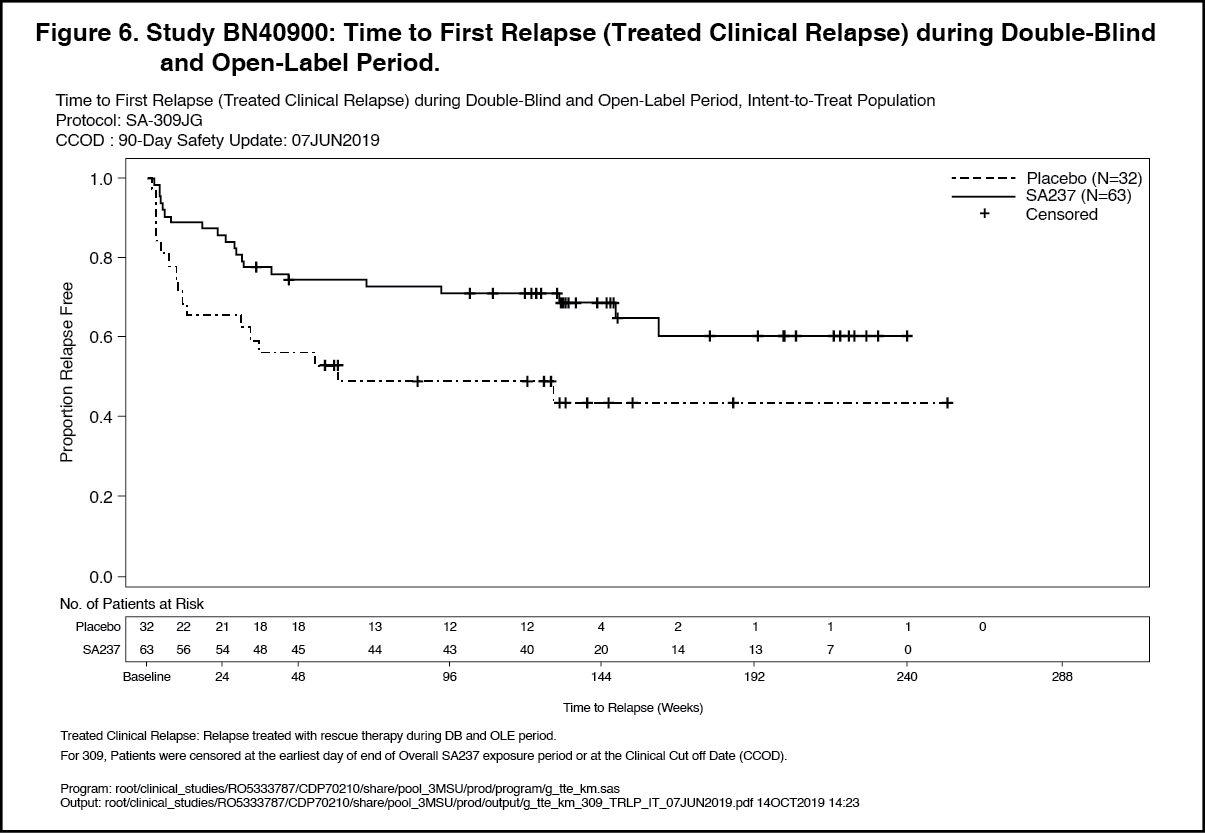 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image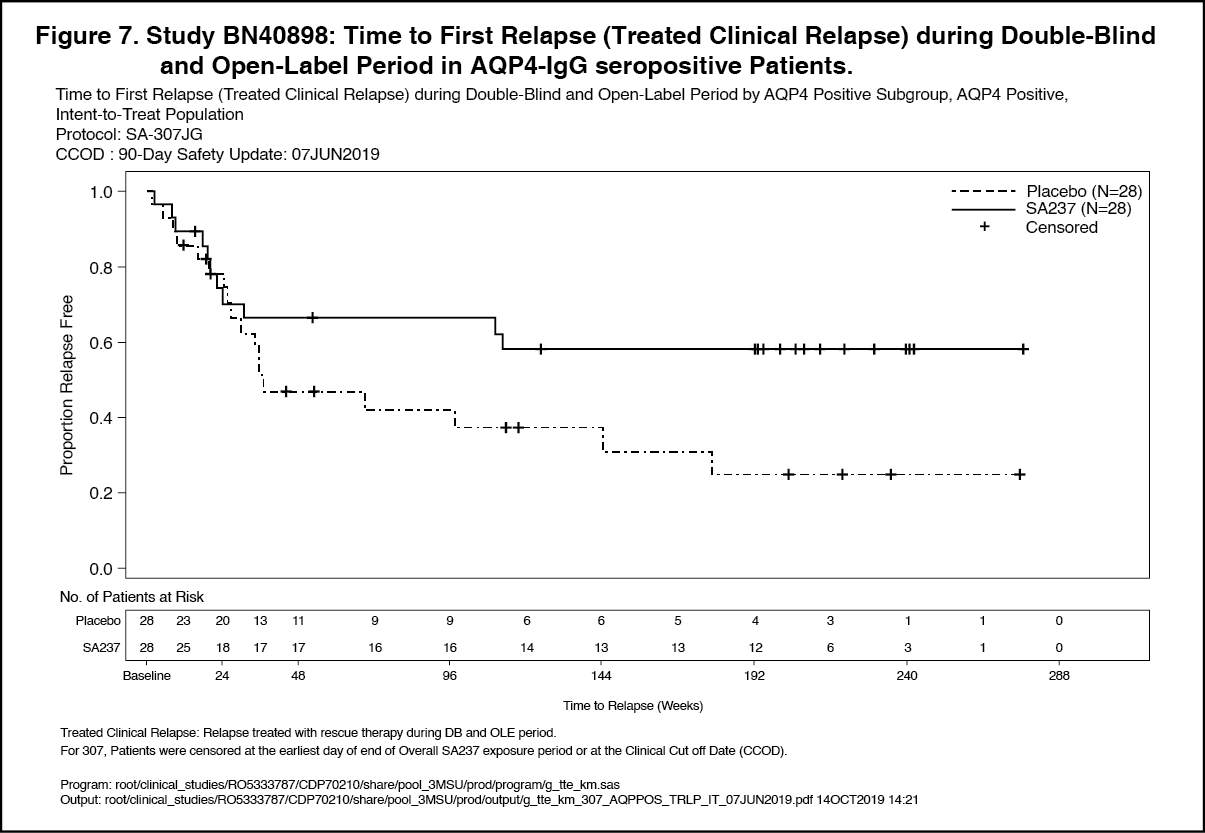 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image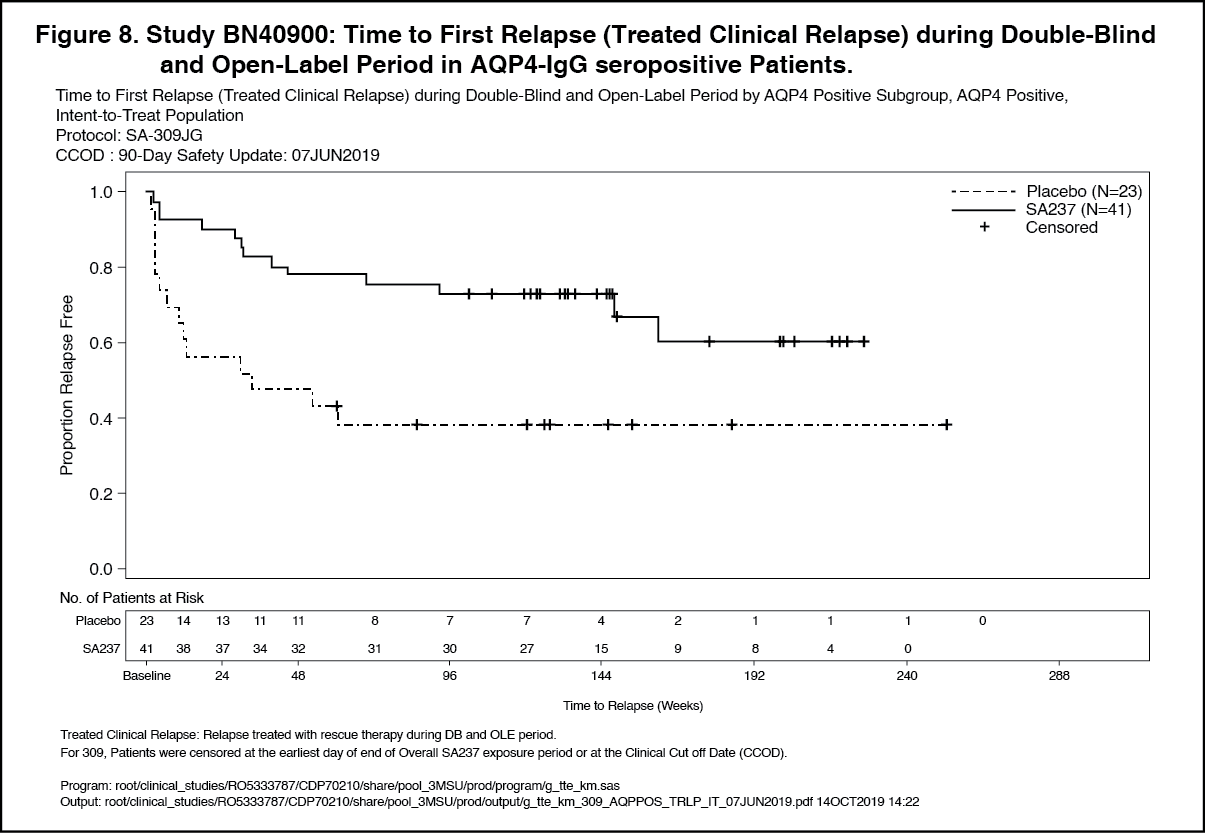 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageBaseline Characteristics and Efficacy in Adolescent Patients (Study BN40898): The mean age of the 7 adolescent patients enrolled during the double-blind period of study BN40898 was 15.4 years and the median body weight was 79.6 kg. The majority of the adolescent patients were females (n=6). Four patients were White, 2 patients were Black/African American, and 1 patient was Asian. Three out of 7 (42.9%) adolescent patients were AQP4-IgG seropositive at screening (2 in the placebo group and 1 in the Enspryng group). During the DB period, 1 of 3 adolescents in the placebo group and 1 of 4 adolescents in the Enspryng group experienced an adjudicated relapse. Due to the small sample size, the hazard ratio for the primary endpoint of time to first adjudicated relapse in this subgroup was not calculated.
Immunogenicity: In phase III Study BN40898 (combination with IST) and in phase III study BN40900 (monotherapy), anti-drug-antibodies (ADAs) were observed in 41% and 71% of patients receiving Enspryng in the double-blind period, respectively. The ability of these ADAs to neutralize Enspryng binding is unknown.
Exposure was lower in ADA positive patients, however there was no impact of ADAs on safety and no clear impact on efficacy nor pharmacodynamic markers indicative of target engagement.
Treatment with satralizumab led to a similar reduction in the risk of experiencing an adjudicated relapse in patients in the phase III studies despite different ADA rates between those studies. Patients with higher bodyweight and lower exposure were more likely to develop ADAs (irrespective of background treatment with IST), however treatment effect was comparable in all bodyweight groups when used either in combination with IST, or as monotherapy. The recommended dose is appropriate for all patients, and neither dose interruption nor modification is warranted in patients who develop ADAs.
Pharmacokinetics: The pharmacokinetics of Enspryng have been characterized both in Japanese and Caucasian healthy volunteers, and in NMO and NMOSD patients. The pharmacokinetics in NMO and NMOSD patients using the recommended dose were characterized using population pharmacokinetic analysis methods based on a database of 154 patients.
The concentration-time course of Enspryng in patients with NMO or NMOSD was accurately described by a two-compartment population PK model with parallel linear and target-mediated (Michaelis-Menten) elimination and first-order SC absorption. Enspryng clearance and volume parameters allometrically scaled by body weight (through power function with the fixed power coefficient of 0.75 and 1 for clearance and volume parameters, respectively). Bodyweight was shown to be a significant covariate, with clearance and Vc for patients weighing 123 kg (97.5th percentile of the weight distribution) increased by 71.3% and 105%, respectively, compared to a 60 kg patient.
Steady state pharmacokinetics were achieved after the loading period (8 weeks) for Cmin, Cmax and AUC as follows (mean (±SD)): Cmin: 19.7 (12.2) mcg/mL, Cmax: 31.5 (14.9) mcg/mL and AUC: 737 (386) mcg.mL/day. Pharmacokinetics were not impacted by background immunotherapy (see Interactions).
Absorption: The absorption rate constant of Enspryng was 0.251 l/day (95% CI: 0.216-0.285) equating to an absorption half-life of around 3 days at the recommended dose (see Dosage & Administration). The bioavailability was high (85.4%, 95% CI: 79.5-95.3%).
Distribution: Enspryng undergoes biphasic distribution. The central volume of distribution was 3.46 L (95% CI: 3.21-3.97), the peripheral volume of distribution was 2.07 L (95% CI: 1.78-2.59). The inter-compartmental clearance was 0.336 L/day (95% CI: 0.261-0.443).
Metabolism: The metabolism of Enspryng has not been directly studied, as monoclonal antibodies are cleared principally by catabolism.
Elimination: The total clearance of Enspryng is concentration-dependent. Linear clearance (accounting for approximately half of the total clearance at steady state using the recommended dose in NMO and NMOSD patients) is estimated to be 0.0601 L/day (95% CI: 0.0524-0.0695). The associated terminal t½ is approximately 30 days (range 22-37 days) based on data pooled from the phase 3 studies.
Pharmacokinetics in Special Populations: Population pharmacokinetic analyses in adult patients with NMO or NMOSD showed that age, gender, and race did not meaningfully influence the pharmacokinetics of satralizumab. Although body weight influenced the pharmacokinetics of satralizumab, no dose adjustments are recommended for any of these demographics.
Pediatric Population: Data obtained in 8 adolescent patients [13-17 years] who received the adult dosing regimen show that population PK parameters for satralizumab are not significantly different from those in the adult population.
No dose adjustment is therefore necessary.
Geriatric Population: No dedicated studies have been conducted to investigate the PK of satralizumab in patients >65 years, however patients with NMO or NMOSD between 65 and 74 years were included in the BN40898 and BN40900 clinical studies.
Population PK analyses based on data from in these patients showed that age did not affect the PK of satralizumab.
Renal impairment: No formal study of the effect of renal impairment on the PK of satralizumab has been conducted. However, patients with mild renal impairment (creatinine clearance <80 mL/min and ≥50 mL/min) were included in the BN40898 and BN40900 clinical studies. As anticipated based on the known mechanisms of clearance for satralizumab, the PK in these patients was not impacted and therefore no dose adjustment is required.
Hepatic impairment: No formal study of the effect of hepatic impairment on the PK of satralizumab has been conducted.
Toxicology: Preclinical safety data: Carcinogenicity: No rodent carcinogenicity studies have been performed to establish the carcinogenic potential of satralizumab. Proliferate lesions have not been observed in a chronic cynomolgus monkey 6-month toxicity study.
Genotoxicity: No studies have been performed to establish the mutagenic potential of satralizumab.
Antibodies are not expected to cause effects on the DNA.
Impairment of Fertility: No effects on male or female reproductive organs were seen with chronic treatment of satralizumab in monkeys.
Reproductive Toxicity: Pre-natal treatment until delivery with up to 50 mg/kg/week satralizumab in pregnant monkeys and postnatal exposure in their offspring did not elicit any adverse effects on maternal animals, fetal development, pregnancy outcome or infant survival and development including learning ability.
The concentrations of satralizumab in breast milk were very low (<0.9% of the corresponding maternal plasma levels).
Other: Repeat dose toxicity: Nonclinical studies with monkeys, a responder species with cross-reactivity to satralizumab did not reveal special hazards for humans based on safety pharmacology, acute and repeated dose toxicity endpoints. When up to 50 mg/kg satralizumab was administered to cynomolgus monkeys once a week in 4- and 26-week repeated-dose SC toxicity studies, no toxicity changes considered to be caused by drug administration were observed. The only relevant change in these studies was increase in blood IL-6 level, which was considered to be the result of the pharmacological action (IL-6R neutralizing action) of satralizumab, and not associated with any adverse findings. Treatment with satralizumab elicited an immune response with anti-drug antibodies in most of the treated animals, which was, however, not affecting the pharmacological response and did not result in any adverse events.
Local tolerance: The SC injection of the clinical formulation of satralizumab did not elicit any adverse reaction at the administration site in monkeys.
Tissue cross-reactivity: Tissue cross-reactivity detected with satralizumab in monkey and human tissues reflects the sites of IL-6R expression. No relevant tissue cross-reactivity was detected in other tissues.
Cytokine release syndrome: Based on in vitro studies with human blood, the risk of the release of proinflammatory cytokines with satralizumab is considered low in terms of incidence and increase in cytokines.