Pharmacology: Mechanism of Action: EVENITY inhibits the action of sclerostin, a regulatory factor in bone metabolism. EVENITY increases bone formation and, to a lesser extent, decreases bone resorption. Animal studies showed that romosozumab stimulates new bone formation on trabecular and endocortical bone surfaces by stimulating osteoblastic activity resulting in increases in trabecular and cortical bone mass and improvements in bone structure and strength [see Nonclinical Toxicology and Clinical Studies as follows].
Pharmacodynamics: In postmenopausal women with osteoporosis, EVENITY increased the bone formation marker procollagen type 1 N-telopeptide (P1NP) with a peak increase from baseline of approximately 145% compared to placebo 2 weeks after initiating treatment, followed by a return to concentrations seen with placebo at month 9 and a decline from baseline to approximately 15% below the concentration change seen with placebo at month 12.
EVENITY decreased the bone resorption marker type 1 collagen C-telopeptide (CTX) with a maximal reduction from baseline of approximately 55% compared to placebo 2 weeks after initiating treatment.
CTX remained below concentrations seen with placebo and was approximately 25% below the concentration change seen with placebo at month 12.
After discontinuation of EVENITY, P1NP levels returned to baseline within 12 months; CTX increased above baseline levels within 3 months and returned toward baseline levels by month 12.
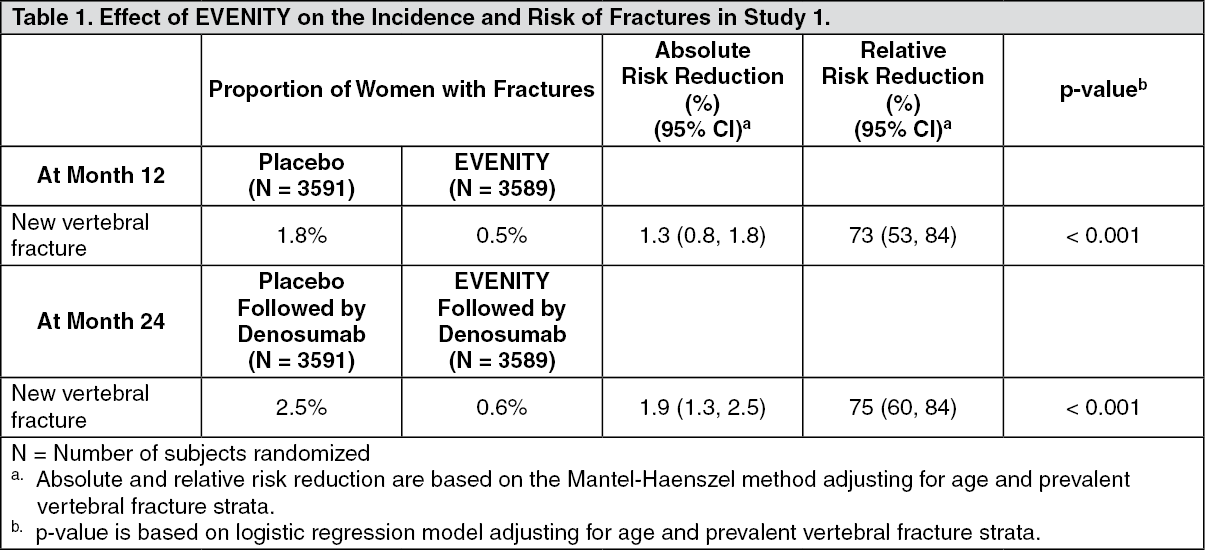
Clinical Studies:Treatment of Osteoporosis in Postmenopausal Women: Study 1 (NCT01575834) was a randomized, double-blind, placebo-controlled study of postmenopausal women aged 55 to 90 years (mean age of 71 years) with bone mineral density (BMD) T-score less than or equal to -2.5 at the total hip or femoral neck. Women were randomized to receive subcutaneous injections of either EVENITY (N = 3589) or placebo (N = 3591) for 12 months. At baseline, 18% of women had a vertebral fracture. After the 12-month treatment period, women in both arms transitioned to open-label anti-resorptive therapy (denosumab) for 12 months while remaining blinded to their initial treatment. Women received 500 to 1000 mg calcium and 600 to 800 international units vitamin D supplementation daily. The coprimary efficacy endpoints were new vertebral fracture at month 12 and month 24.
Effect on Fractures: EVENITY significantly reduced the incidence of new vertebral fractures through month 12 compared to placebo. In addition, the significant reduction in fracture risk persisted through the second year in women who received EVENITY during the first year and transitioned to denosumab compared to those who transitioned from placebo to denosumab (see Table 1).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
EVENITY significantly reduced the incidence of clinical fracture (a composite endpoint of symptomatic vertebral fracture and nonvertebral fracture) at 12 months. However, 88% of these clinical fractures were nonvertebral fractures and the incidence of nonvertebral fractures was not statistically significantly different when comparing EVENITY-treated women to placebo-treated women at month 12 or month 24.
Effect on BMD: EVENITY significantly increased BMD at the lumbar spine, total hip, and femoral neck compared with placebo at month 12. The treatment differences in BMD were 12.7% at the lumbar spine, 5.8% at the total hip, and 5.2% at the femoral neck.
Following the transition from EVENITY to denosumab at month 12, BMD continued to increase through month 24. In patients who transitioned from placebo to denosumab, BMD also increased with denosumab use. The differences in BMD achieved at month 12 between EVENITY and placebo patients were overall maintained at month 24, when comparing patients who transitioned from EVENITY to denosumab to those who transitioned from placebo to denosumab. There was no evidence of differences in effects on BMD at the lumbar spine or total hip across subgroups defined by baseline age, baseline BMD, or geographic region.
After EVENITY discontinuation, BMD returns to approximately baseline levels within 12 months in the absence of follow-on anti-resorptive therapy [see Indications/Uses].
Bone Histology and Histomorphometry: A total of 154 transiliac crest bone biopsy specimens were obtained from 139 postmenopausal women with osteoporosis at month 2, month 12, and/or month 24. All of these biopsies were adequate for qualitative histology and 138 (90%) were adequate for full quantitative histomorphometry assessment.
Qualitative histology assessments from women treated with EVENITY showed normal bone architecture and quality at all time points. There was no evidence of woven bone, mineralization defects, or marrow fibrosis.
Histomorphometry assessments on biopsies at months 2 and 12 compared the effect of EVENITY with placebo (15 specimens at month 2 and 39 specimens at month 12 in the EVENITY group, 14 specimens at month 2 and 31 specimens at month 12 in the placebo group). At month 2 in women treated with EVENITY, histomorphometric indices of bone formation at trabecular and endocortical surfaces were increased. These effects on bone formation were accompanied by a decrease in indices of bone resorption.
At month 12, both bone formation and resorption indices were decreased with EVENITY, while bone volume, and trabecular and cortical thickness were increased.
Study 2 (NCT01631214) was a randomized, double-blind, alendronate-controlled study of postmenopausal women aged 55 to 90 years (mean age of 74 years) with BMD T-score less than or equal to -2.5 at the total hip or femoral neck and either one moderate or severe vertebral fracture or two mild vertebral fractures, or BMD T-score less than or equal to -2.0 at the total hip or femoral neck and either two moderate or severe vertebral fractures or a history of a proximal femur fracture. Women were randomized (1:1) to receive either monthly subcutaneous injections of EVENITY (N = 2046) or oral alendronate 70 mg weekly (N = 2047) for 12 months, with 500 to 1000 mg calcium and 600 to 800 international units vitamin D supplementation daily. After the 12-month treatment period, women in both arms transitioned to open-label alendronate 70 mg weekly while remaining blinded to their initial treatment.
This was an event-driven trial. The coprimary efficacy endpoints were the incidence of morphometric vertebral fracture at 24 months and time to the first clinical fracture through the primary analysis period, which ended when at least 330 subjects had a clinical fracture and all subjects had completed the 24-month visit. Clinical fracture was a composite endpoint of nonvertebral fracture and symptomatic vertebral fracture.
Effect on Fractures: EVENITY significantly reduced the incidence of new vertebral fracture at 24 months (see Table 2).
Click on icon to see table/diagram/image
EVENITY significantly reduced the risk of clinical fracture through the end of the primary analysis period (see Table 3). This was an event-driven trial and the duration of follow-up varied across subjects.
The median duration of subject follow-up for the primary analysis period was 33 months. Subjects with nonvertebral fracture comprised 83% of the subjects with clinical fracture during the primary analysis period. (See Table 3.)
Click on icon to see table/diagram/image
EVENITY followed by alendronate also significantly reduced the risk of nonvertebral fracture through the primary analysis period (with a median follow-up of 33 months), with a hazard ratio of 0.81 (95% CI: 0.66, 0.99; p = 0.04) compared to alendronate alone.
Effect on Bone Mineral Density (BMD): EVENITY significantly increased BMD at the lumbar spine, total hip, and femoral neck compared with alendronate at month 12. The treatment differences in BMD were 8.7% at the lumbar spine, 3.3% at the total hip, and 3.2% at the femoral neck.
Twelve months of treatment with EVENITY followed by 12 months of treatment with alendronate significantly increased BMD compared with alendronate alone. The BMD increase with EVENITY over alendronate observed at month 12 was maintained at month 24. The treatment differences in BMD at month 24 were 8.1% at the lumbar spine, 3.8% at the total hip, and 3.8% at the femoral neck.
There was no evidence of differences in effects on BMD at the lumbar spine or total hip across subgroups defined by baseline age, baseline BMD, or geographic region.
Pharmacokinetics: Administration of a single dose of 210 mg EVENITY in healthy volunteers resulted in a mean (standard deviation [SD]) maximum romosozumab serum concentration (C
max) of 22.2 (5.8) mcg/mL and a mean (SD) AUC of 389 (127) mcg*day/mL. Steady-state concentrations were achieved by month 3 following the monthly administration of 210 mg to postmenopausal women. The mean trough serum romosozumab concentrations at months 3, 6, 9, and 12 ranged from 8 to 13 mcg/mL.
Romosozumab exhibited nonlinear pharmacokinetics with exposure increasing greater than dose proportionally (e.g., 550-fold increase in mean AUC
inf for the 100-fold increase in subcutaneous doses ranging from 0.1 to 10 mg/kg [0.03 to 3.3 times the approved recommended dosage for a 70 kg woman).
Absorption: The median time to maximum romosozumab concentration (T
max) is 5 days (range: 2 to 7 days).
Distribution: The estimated volume of distribution at steady-state is approximately 3.92 L.
Elimination: Romosozumab exhibited nonlinear pharmacokinetics with the clearance of romosozumab decreasing as the dose increased. The estimated mean systemic clearance (CL/F) of romosozumab was 0.38 mL/hr/kg, following a single subcutaneous administration of 3 mg/kg (the approved recommended dosage for a 70 kg woman). The mean effective was t
½ 12.8 days after 3 doses of 3 mg/kg (the approved recommended dosage for a 70 kg woman) every 4 weeks.
Metabolism: The metabolic pathway of romosozumab has not been characterized. As a humanized IgG2 monoclonal antibody, romosozumab is expected to be degraded into small peptides and amino acids via catabolic pathways in a manner similar to endogenous IgG.
Anti-Product Antibody Formation Affecting Pharmacokinetics: Development of anti-romosozumab antibodies was associated with reduced serum romosozumab concentrations. The presence of anti-romosozumab antibodies led to decreased mean romosozumab concentrations up to 22%. The presence of neutralizing antibodies led to decreased mean romosozumab concentrations up to 63% [see Adverse Reactions].
Specific Populations: No clinically significant differences in the pharmacokinetics of romosozumab were observed based on age (20-89 years), sex, race, disease state (low bone mass or osteoporosis), prior exposure to alendronate, or renal impairment including end-stage renal disease (ESRD) requiring dialysis. The effect of ESRD not requiring dialysis on the pharmacokinetics of romosozumab is unknown.
Body Weight: The exposure of romosozumab decreases with increasing body weight.
Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: Carcinogenicity: In a rat carcinogenicity study, once-weekly romosozumab doses of 3, 10 or 50 mg/kg were administered by subcutaneous injection to Sprague-Dawley rats from 8 weeks up to 98 weeks of age, resulting in systemic exposures that were up to 19 times the systemic exposure observed in humans following a monthly subcutaneous dose of 210 mg EVENITY (based on AUC comparison). Romosozumab caused a dose-dependent increase in bone mass with trabecular and cortical bone thickening at all doses. There were no effects of romosozumab on mortality and romosozumab did not cause significant increases in tumor incidence in male or female rats.
Mutagenicity: Mutagenesis has not been evaluated, as monoclonal antibodies are not expected to alter DNA or chromosomes.
Impairment of Fertility: No effects on fertility were observed in male and female rats given subcutaneous romosozumab doses up to 300 mg/kg (up to 56 times the systemic exposure observed in humans following a monthly subcutaneous dose of 210 mg EVENITY, based on AUC comparison). No effects were noted in reproductive organs in rats and cynomolgus monkeys dosed subcutaneously for 6 months with weekly doses up to 100 mg/kg (exposures up to 38 and 93 times, respectively, the systemic exposure observed in humans administered monthly subcutaneous doses of 210 mg based on AUC comparison).
Animal Toxicology and Pharmacology: No adverse effects were noted in rats and monkeys after 26 once-weekly subcutaneous romosozumab doses up to 100 mg/kg, equivalent to systemic exposures of 38 and 93 times, respectively, the systemic exposure observed in humans following a monthly subcutaneous dose of 210 mg EVENITY (based on AUC comparison).
Bone safety studies of up to 12-month duration were conducted in ovariectomized rats and monkeys with once-weekly romosozumab doses yielding exposures ranging from 1 to 22 times the systemic exposure in humans given monthly doses of 210 mg, based on AUC comparison. Romosozumab increased bone mass and improved cancellous bone microarchitecture and cortical bone geometry by increasing bone formation on periosteal, endocortical, and trabecular surfaces, and decreasing bone resorption on trabecular and endocortical surfaces. The increases in bone mass were significantly correlated with increases in bone strength. In rats and monkeys, bone quality was maintained at all skeletal sites at doses ranging from 1 to 22 times human exposure, and slightly improved in vertebrae at 19 to 22 times human exposure. There was no evidence of mineralization defects, osteoid accumulation, or woven bone formation.



 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image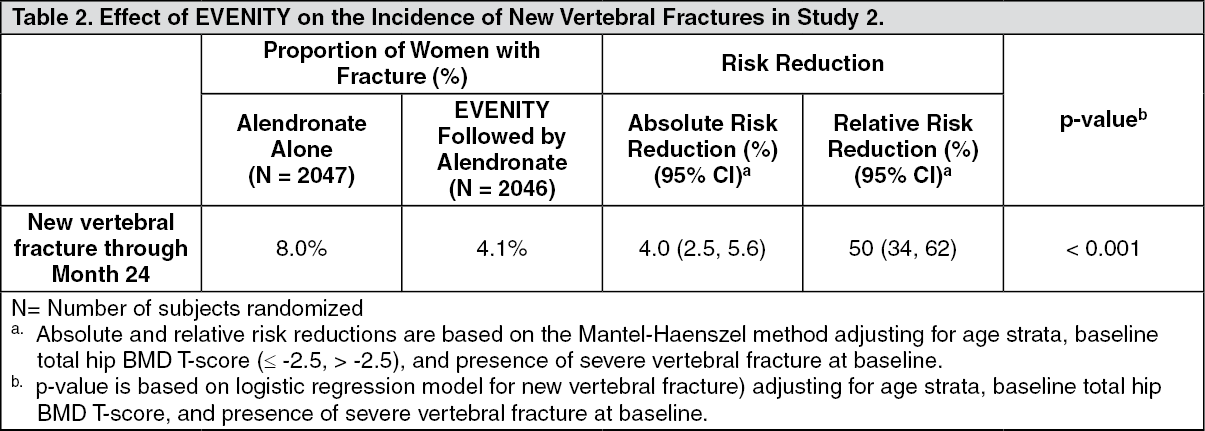 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image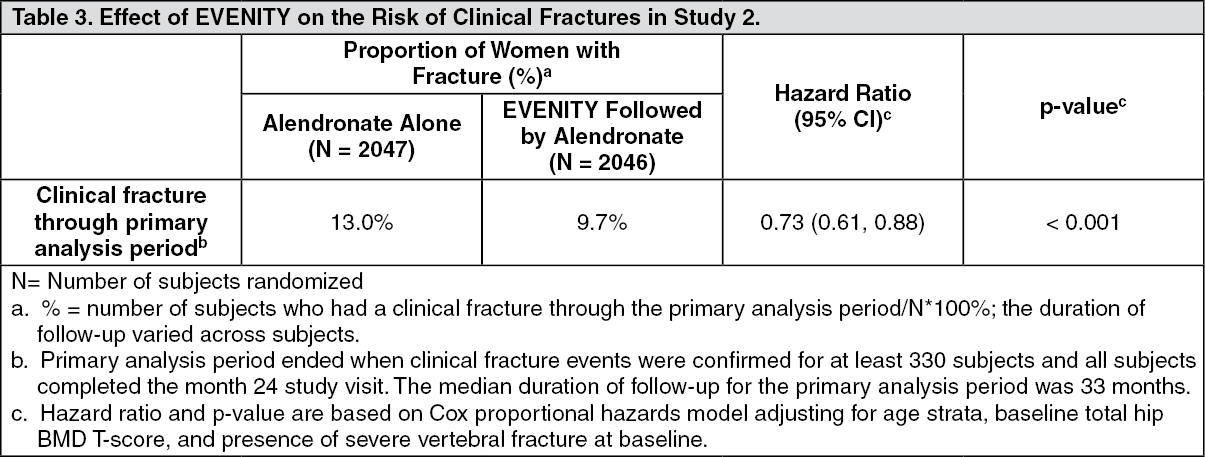 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image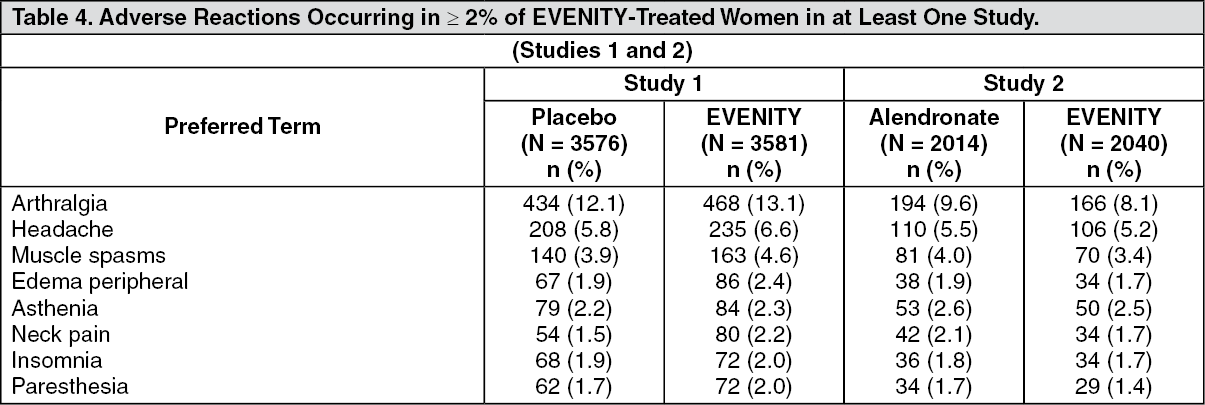 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
