


 Sign Out
Sign Out

Pharmacology: Mechanism of action: Ruxolitinib is a selective inhibitor of the Janus Associated Kinases (JAKs) JAK1 and JAK2 (IC50 values of 3.3 nM and 2.8 nM for JAK1 and JAK2 enzymes, respectively). These mediate the signaling of a number of cytokines and growth factors that are important for hematopoiesis and immune function. JAK signaling involves recruitment of signal transducers and activators of transcription (STATs) to cytokine receptors, activation, and subsequent localization of STATs to the nucleus leading to modulation of gene expression. Dysregulation of the JAK-STAT pathway has been associated with several cancers and increased proliferation and survival of malignant cells.
MF and PV are myeloproliferative neoplasms (MPN) known to be associated with dysregulated JAK1 and JAK2 signaling. The basis for the dysregulation is believed to include high levels of circulating cytokines that activate the JAK-STAT pathway, gain-of function mutations such as JAK2V617F, and silencing of negative regulatory mechanisms. MF patients exhibit dysregulated JAK signaling regardless of JAK2V617F mutation status. Activating mutations in JAK2 (V617F orexon 12) are found in >95% of PV patients.
Ruxolitinib inhibits JAK-STAT signaling and cell proliferation of cytokine-dependent cellular models of hematological malignancies, as well as of Ba/F3 cells rendered cytokine-independent by expressing the JAK2V617F mutated protein, with IC50's ranging from 80-320 nM. In a mouse model of JAK2V617F-positive MPN, oral administration of ruxolitinib prevented splenomegaly, preferentially decreased JAK2V617F mutant cells in the spleen, decreased circulating inflammatory cytokines (e.g., TNF-alpha, IL-6) and resulted in significantly prolonged survival in the mice at doses that did not cause myelosuppressive effects.
JAK-STAT signaling pathways play a role in regulating the development, proliferation, and activation of several immune cell types important for GvHD pathogenesis. In a mouse model of acute GvHD, oral administration of ruxolitinib was associated with decreased expression of inflammatory cytokines in colon homogenates and reduced immune-cell infiltration in the colon.
Pharmacodynamics: Ruxolitinib inhibits cytokine induced STAT3 phosphorylation in whole blood from healthy subjects and MF and PV patients. Ruxolitinib resulted in maximal inhibition of STAT3 phosphorylation 2 hours after dosing which returned to near baseline by 8 hours in both healthy subjects and MF patients, indicating no accumulation of either parent or active metabolites.
Baseline elevations in inflammatory markers associated with constitutional symptoms such as TNF-alpha, IL-6, and CRP in patients with MF were decreased following treatment with Jakavi. Patients with MF did not become refractory to the pharmacodynamic effects of Jakavi treatment over time. Similarly, patients with PV also presented with baseline elevations in inflammatory markers and these markers were decreased following treatment with Jakavi.
In a thorough QT study in healthy subjects, there was no indication of a QT/QTc prolonging effect of ruxolitinib in single doses up to a supratherapeutic dose of 200 mg indicating that ruxolitinib has no effect on cardiac repolarization.
Clinical Studies: Myelofibrosis: Two randomized Phase 3 studies (COMFORT-I and COMFORT-II) were conducted in patients with MF (PMF, PPV-MF or PET-MF). In both studies, patients had palpable splenomegaly at least 5 cm below the costal margin and risk category of intermediate 2 (2 prognostic factors) or high risk (3 or more prognostic factors) based on the International Working Group Consensus Criteria (IWG). The prognostic factors that comprise the IWG criteria consist of age >65 years, presence of constitutional symptoms (weight loss, fever, night sweats), anemia (hemoglobin <10 g/dL), leukocytosis (history of WBC >25 X 109/L) and circulating blasts ≥1%. The starting dose of Jakavi was based on platelet count. Patients with a platelet count between 100,000 and 200,000/mm3 were started on Jakavi 15 mg twice daily and patients with a platelet count >200,000/mm3 were started on Jakavi 20 mg twice daily. Of the 301 patients, 111 (36.9%) had a baseline platelet count between 100,000 and 200,000/mm3, and 190 (63.1%) had a baseline platelet count >200,000/mm3 [Table 6]. Patients with platelet counts ≤100,000/mm3 were not eligible in COMFORT studies. Maximum safe starting dose (MSSD) for patients with baseline platelet counts ≥50,000 and <100,000/mm3 was confirmed as 10 mg twice daily in EXPAND, a Phase Ib, open label, dose-finding study in patients with PMF, PPV-MF or PET-MF. In COMFORT studies, doses were individualized based upon tolerability and efficacy with maximum doses of 20 mg twice daily for patients with platelet counts between 100,000 to ≤125,000/mm3, of 10 mg twice daily for patients with platelet counts between 75,000 to ≤100,000/mm3, and of 5 mg twice daily for patients with platelet counts between 50,000 to ≤75,000/mm3.
COMFORT-I was a double-blind, randomized, placebo-controlled study in 309 patients who were refractory to or were not candidates for available therapy. Patients were dosed with Jakavi or matching placebo. The primary efficacy endpoint was proportion of subjects achieving ≥35% reduction from baseline in spleen volume at Week 24 as measured by MRI or CT.
Secondary endpoints included duration of maintenance of a ≥35% reduction from baseline in spleen volume, proportion of patients who had a ≥50% reduction in total symptom score from baseline to Week 24 as measured by the modified Myelofibrosis Symptom Assessment Form (MFSAF) v2.0 diary, change in total symptom score from baseline to Week 24 as measured by the modified MFSAF v2.0 diary and overall survival.
COMFORT-II was an open-label, randomized study in 219 patients. Patients were randomized 2:1 to Jakavi versus BAT. BAT was selected by the investigator on a patient-by-patient basis. In the BAT arm, 47% of patients received hydroxyurea and 16% of patients received glucocorticoids. The primary efficacy endpoint was proportion of patients achieving ≥35% reduction from baseline in spleen volume at Week 48 as measured by MRI or CT.
A secondary endpoint in COMFORT-II was the proportion of patients achieving a ≥35% reduction of spleen volume measured by MRI or CT from baseline to Week 24. Duration of maintenance of a ≥35% reduction from baseline in responding patients was also a secondary endpoint.
In COMFORT-I, patient baseline demographics and disease characteristics were comparable between the treatment arms. The median age was 68 years with 61% of patients older than 65 years and 54% male. Fifty percent (50%) of patients had PMF, 31% had PPV-MF and 18% had PET-MF. Twenty-one percent (21%) of patients had red blood transfusions within 8 weeks of enrollment in the study. The median platelet count was 251,000/mm3. Seventy-six percent (76%) of patients had the mutation encoding the V617F substitution present in the JAK protein. Patients had a median palpable spleen length of 16 cm. At baseline 37.4% of the patients in the Jakavi arm had Grade 1 anemia, 31.6% Grade 2 and 4.5% Grade 3, while in the placebo arm 35.8% had Grade 1, 35.1% Grade 2, 4.6% Grade 3, and 0.7% Grade 4. Grade 1 thrombocytopenia was found in 12.9% of patients in the Jakavi arm and 13.2% in the placebo arm.
In COMFORT-II, patient baseline demographics and disease characteristics were comparable between the treatment arms. The median age was 66 years with 52% of patients older than 65 years and 57% male. Fifty-three percent (53%) of the patients had PMF, 31% had PPV-MF, and 16% had PET-MF. Nineteen percent (19%) of the patients were considered transfusion dependent at baseline. Patients had a median palpable spleen length of 15 cm.
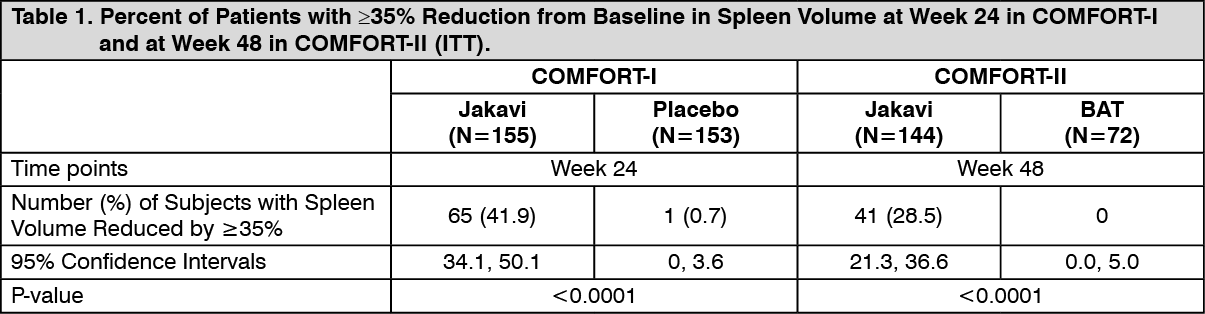
At baseline 34.2% of the patients in the Jakavi arm had Grade 1 anemia, 28.8% Grade 2, and 7.5% Grade 3, while in the BAT arm 37% had Grade 1, 27.4% Grade 2, 13.7% Grade 3, and 1.4% Grade 4. Thrombocytopenia of Grade 1 was found in 8.2% of the patients in the Jakavi arm, and 9.6% in the BAT arm. Efficacy analyses of the primary endpoint in COMFORT-I and COMFORT-II are presented in Table 1. A significantly larger proportion of patients in the Jakavi arm achieved a ≥35% reduction in spleen volume from baseline in both studies compared to placebo in COMFORT-I and BAT in COMFORT-II. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageIn COMFORT-I, 41.9% of patients in the Jakavi arm achieved a ≥35% reduction in spleen volume from baseline compared with 0.7% in the placebo arm at Week 24. A similar proportion of patients in the Jakavi arm achieved a ≥50% reduction in palpable spleen length.
In COMFORT-II, 28.5% of patients in the Jakavi arm achieved a ≥35% reduction in spleen volume from baseline compared with none (0%) in the BAT arm at Week 48. A secondary endpoint was the proportion of patients achieving a ≥35% reduction of spleen volume at Week 24. A significantly larger proportion of patients in the Jakavi group 46 patients (31.9%) achieved a ≥35% reduction in spleen volume from baseline compared to no (0%) patients in the BAT arm (p-value <0.0001).
A significantly higher proportion of patients in the Jakavi arm achieved a ≥35% reduction from baseline in spleen volume regardless of the presence or absence of the JAK2V617F mutation or the disease subtype (PMF, PPV-MF, PET-MF).
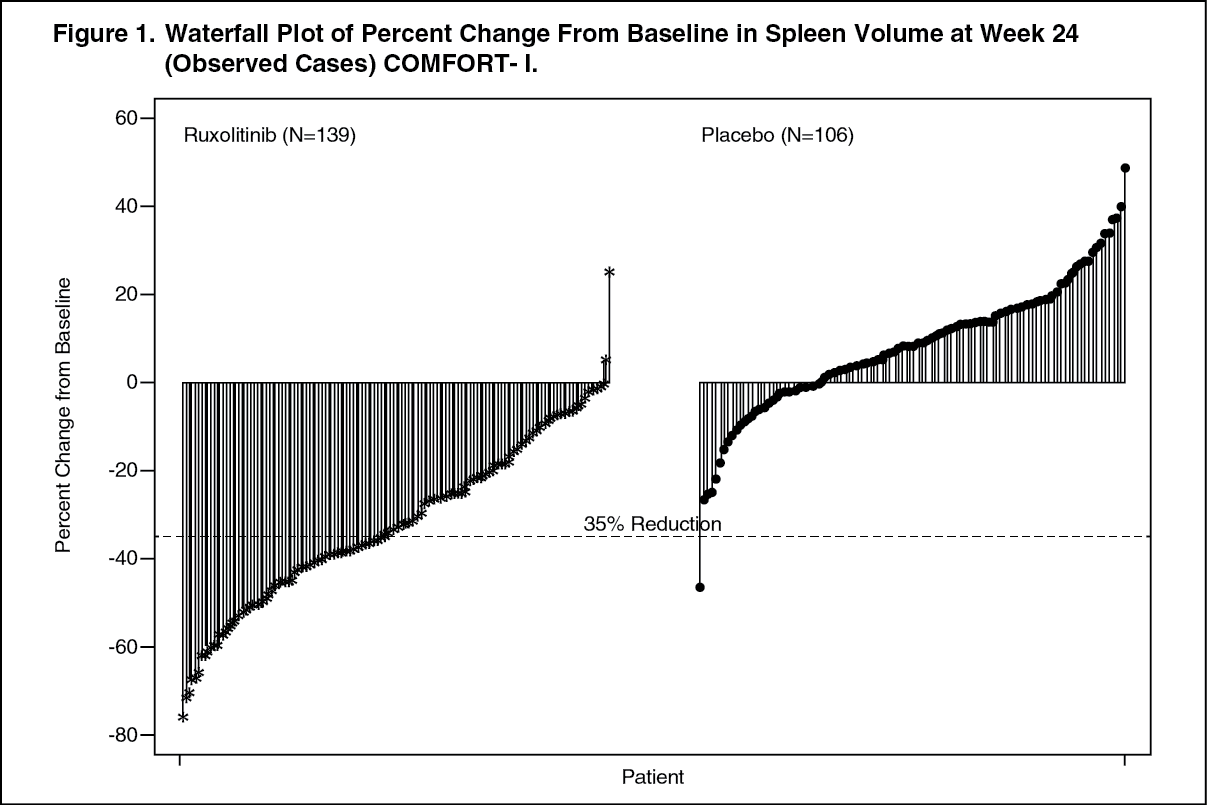
Figure 1 shows a waterfall plot of the percent change from baseline in spleen volume at Week 24 in COMFORT-I. Among the 139 patients in the Jakavi arm who had both baseline and Week 24 spleen volume evaluations, all but two patients had some level of reduction in spleen volume at Week 24, with a median reduction of 33%. Among the 106 patients in the placebo arm who had both baseline and Week 24 spleen volume evaluations, there was a median increase of 8.5%. (See Figure 1.)
 Click on icon to see table/diagram/image
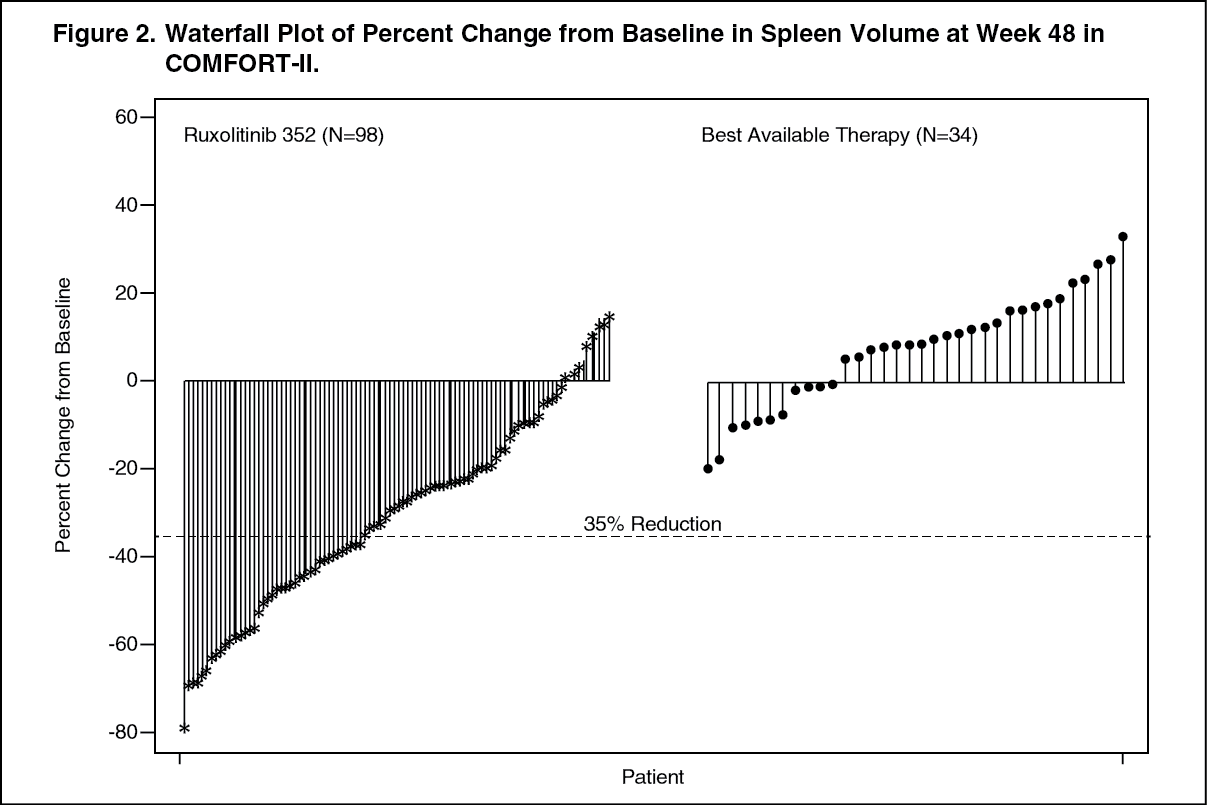
Click on icon to see table/diagram/imageFigure 2 shows a waterfall plot of the percent change from baseline in spleen volume at Week 48 in COMFORT-II. Among the 98 patients in the Jakavi arm who had both baseline and Week 48 spleen volume evaluations, the median reduction in spleen volume at Week 48 was 28%. Among the 34 patients in the BAT arm who had both baseline and Week 48 spleen volume evaluations, there was a median increase of 8.5%. (See Figure 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe probability of duration from 1st ≥35% reduction of spleen volume to 25% increase from nadir and loss of response in COMFORT-I and COMFORT-II is shown in Table 2. (See Table 2.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageAmong the 80 patients that showed a ≥35% reduction at any time point in COMFORT-I and of the 69 patients in COMFORT-II, the probability that a patient would maintain a response to Jakavi for at least 24 weeks was 89% and 87% in COMFORT-I and COMFORT-II respectively and the probability of maintaining a response for at least 48 weeks was 52% in COMFORT-II.
Jakavi improved MF-related symptoms and quality of life (QOL) in patients with PMF, PPV-MF and PET-MF. In COMFORT-I symptoms of MF were captured using the modified MFSAF diary v2.0 as an electronic diary, which patients completed daily. The change from baseline in the Week 24 total score was a secondary endpoint in this study. Significantly larger proportion of patients in the Jakavi arm achieved a ≥50% improvement from baseline in the Week 24 total symptom score compared with the placebo arm (45.9% and 5.3%, respectively, p <0.0001 using the Chi-Squared test). An improvement in overall quality of life was measured by the European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire (QLQ)-C30 in COMFORT-I and COMFORT-II. COMFORT-I compared Jakavi to placebo at 24 weeks and COMFORT-II compared Jakavi to BAT at 48 weeks. At baseline for both studies, EORTC QLQ-C30 individual subscale scores for the Jakavi and comparator arms were similar. At Week 24 in COMFORT-I, the Jakavi arm showed significant improvement in the global health status/QOL of the EORTC QLQ-C30 compared with the placebo arm (mean change of +12.3 and -3.4 for Jakavi and placebo, respectively, p <0.0001). At week 24 and week 48, the Jakavi arm in COMFORT-II showed a trend towards greater improvement of global health status/QOL compared to BAT, an exploratory endpoint, consistent with the COMFORT-I findings.
In COMFORT-I, after a median follow-up of 34.3 months, the death rate in patients randomized to the Jakavi arm was 27.1% (42 of 155 patients) versus 35.1% (54 of 154) in patients randomized to placebo. There was a 31.3% reduction in the risk of death in the Jakavi arm as compared to placebo (HR: 0.687; 95% CI: 0.459-1.029; p=0.0668). At final analysis, after a median follow up of 61.7 months, the reduction in risk of death was maintained at 30.7% (HR: 0.693; 95% CI: 0.503, 0.956, p=0.025).
In COMFORT-II, after a median follow-up of 34.7 months, the death rate in patients randomized to Jakavi was 19.9% (29 of 146 patients) versus 30.1% (22 of 73 patients) in patients randomized to BAT. There was a 52% reduction in risk of death in the Jakavi arm compared to the BAT arm (HR: 0.48; 95% CI: 0.28-0.85; p=0.009). At final analysis, after a median follow up of 55.9 months, the reduction in risk of death was consistent with COMFORT-I (HR: 0.67, 95% CI: 0.44-1.02, p=0.062).
Polycythemia vera: A randomized, open-label, active-controlled Phase 3 study (RESPONSE) was conducted in 222 patients with PV who were resistant to or intolerant of hydroxyurea. A total of 110 patients were randomized to the Jakavi arm and 112 patients to the BAT arm. The starting dose of Jakavi was 10 mg twice daily. Doses were then adjusted in individual patients based on tolerability and efficacy with a maximum dose of 25 mg twice daily. BAT was selected by the investigator on a patient-by-patient basis and included hydroxyurea (59.5%), interferon/pegylated interferon (11.7%), anagrelide (7.2%), pipobroman (1.8) and observation (15.3%).
Baseline demographics and disease characteristics were comparable between the two treatments arms. The median age was 60 years (range 33 to 90 years). Patients in the Jakavi arm had PV diagnosis for a median of 8.2 years and had previously received hydroxyurea for a median of approximately 3 years. Most patients (>80%) had received at least two phlebotomies in the last 24 weeks prior to screening.
The primary composite endpoint was the proportion of patients achieving both the absence of phlebotomy eligibility (HCT control) and ≥35% reduction in spleen volume from baseline at Week 32. Phlebotomy eligibility was defined as a confirmed HCT >45% that is at least 3 percentage points higher than the HCT obtained at baseline or a confirmed HCT >48%, whichever is lower. Key secondary endpoints included the proportion of patients who achieved the primary endpoint and who remained free from progression at Week 48, and the proportion of patients achieving complete hematological remission at Week 32.
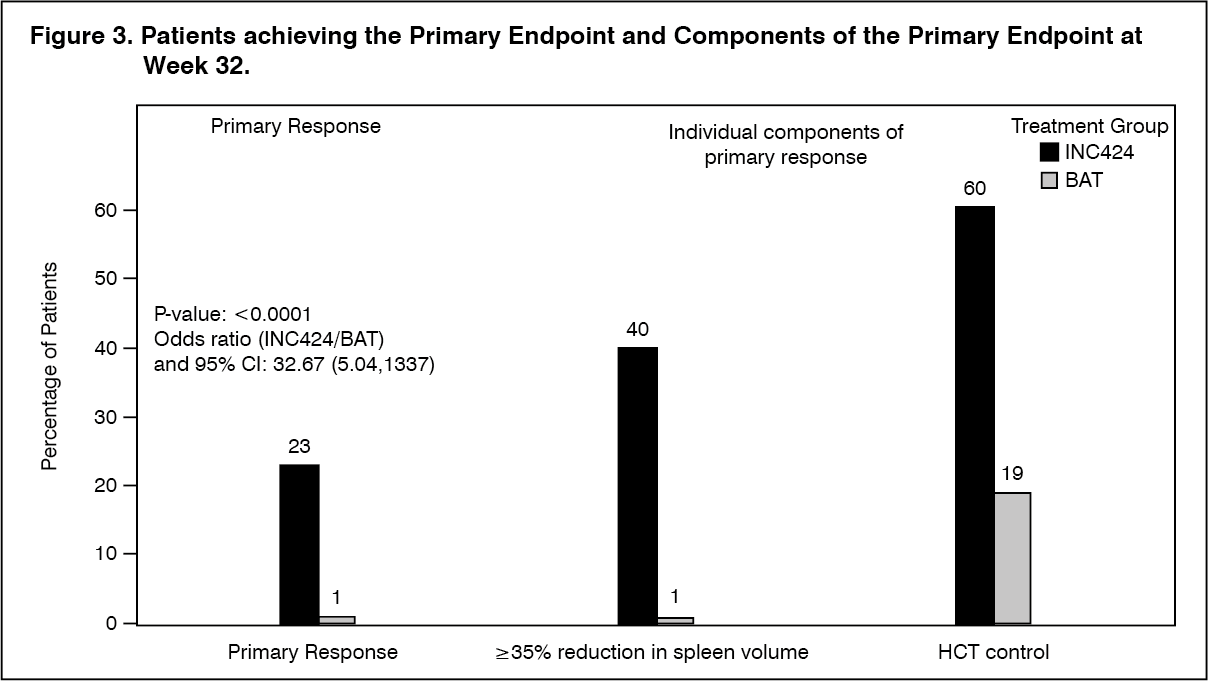
The study met its primary objective and a higher proportion of patients in the Jakavi arm achieved the primary composite endpoint and each of its individual components. Significantly more patients on Jakavi (23%) compared to BAT (0.9%) achieved a primary response (p<0.0001). HCT control was achieved in 60% of patients in the Jakavi arm compared to 18.75% in the BAT arm and ≥35% reduction in spleen volume was achieved in 40% of patients in the Jakavi arm compared to 0.9% in the BAT arm (Figure 3).
Both key secondary endpoints were also met: The proportion of patients achieving a complete hematologic remission was 23.6% on Jakavi compared to 8.0% on BAT (p=0.0013), and the proportion of patients achieving a durable primary response at week 48 was 20% on Jakavi and 0.9% on BAT (p<0.0001). (See Figure 3.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageSymptom burden was assessed using the MPN-Symptoms Assessment Form (SAF) total symptom score (TSS) electronic patient diary consisting of 14 questions. At Week 32, 49% and 64% of patients treated with Jakavi achieved a ≥50% reduction in TSS-14 and TSS-5, respectively, compared to only 5% and 11% of patients on BAT.
Treatment benefit perception was measured by the Patient Global Impression of Change (PGIC) questionnaire. A total of 66% of Jakavi-treated patients compared to 19% in BAT reported an improvement as early as 4 weeks after the start of treatment. Improvement in perception of treatment benefit was also higher in Jakavi-treated patients at Week 32 (78% versus 33%).
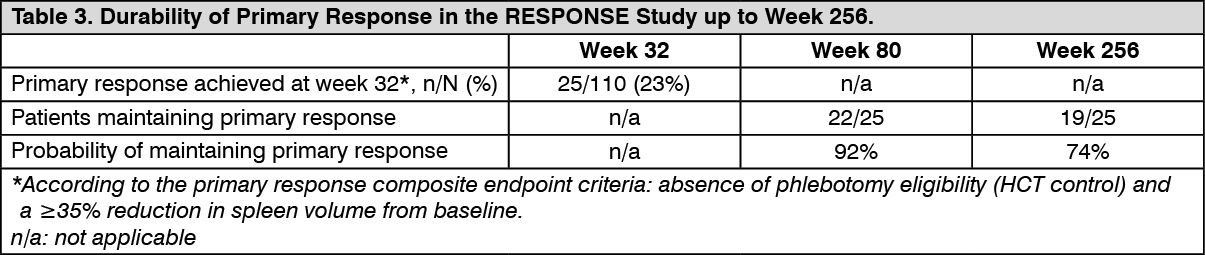
Additional analyses from the RESPONSE study to assess durability of response were conducted at Week 80 and week 256 following randomization. Out of 25 patients who had achieved primary response at week 32, 3 patients had progressed by week 80 and 6 patients by week 256. The probability to have maintained a response from week 32 up to week 80 and week 256 was 92% and 74%, respectively (see Table 3).
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageA second randomized, open label, active-controlled phase IIIb study (RESPONSE-2) was conducted in 149 PV patients who were resistant to or intolerant of hydroxyurea but without palpable splenomegaly. Seventy-four patients were randomized to the Jakavi arm and 75 patients to the BAT arm. The starting dose and dose adjustments of Jakavi and investigator-selected BAT were similar to the RESPONSE study. Baseline demographics and disease characteristics were comparable between the two treatment arms and similar to the patient population of the RESPONSE study. The primary endpoint was the proportion of patients achieving HCT control (absence of phlebotomy eligibility) at Week 28. The key secondary endpoint was the proportion of patients achieving complete hematological remission at Week 28.
RESPONSE-2 met its primary objective with a higher proportion of patients in the Jakavi arm (62.2%) compared to the BAT arm (18.7%) achieving the primary endpoint (p<0.0001). The key secondary endpoint was also met with significantly more patients achieving a complete hematologic remission in the Jakavi arm (23.0%) compared to the BAT arm (5.3%; p=0.0019). At week 28, the proportion of patients achieving a ≥50% reduction in symptom burden as measured by the MPN-SAF total symptom score was 45.3% in the Jakavi arm and 22.7% in the BAT arm.
Graft-versus-host disease: Two randomized phase 3, open-label, multi-center studies investigated Jakavi in patients 12 years of age and older with acute GvHD (REACH2) and chronic GvHD (REACH3) after allogeneic hematopoietic stem cell transplantation (alloSCT) and insufficient response to corticosteroids and other systemic therapies. The starting dose of Jakavi was 10 mg twice daily.
Acute graft-versus-host disease: In REACH2, 309 patients with Grade II to IV corticosteroid-refractory, acute GvHD were randomized 1:1 to Jakavi or BAT. Patients were stratified by severity of acute GvHD at the time of randomization. Corticosteroid refractoriness was determined when patients had progression after at least 3 days, failed to achieve a response after 7 days or failed corticosteroid taper.
BAT was selected by the investigator on a patient-by-patient basis and included anti-thymocyte globulin (ATG), extracorporeal photopheresis (ECP), mesenchymal stromal cells (MSC), low dose methotrexate (MTX), mycophenolate mofetil (MMF), mTOR inhibitors (everolimus or sirolimus), etanercept, or infliximab.
In addition to Jakavi or BAT, patients could have received standard allogeneic stem cell transplantation supportive care including anti-infective medications and transfusion support as well as standard acute GvHD prophylaxis and treatment medications initiated before randomization including systemic corticosteroids and calcineurin inhibitors (CNIs) such as cyclosporine or tacrolimus. Topical or inhaled corticosteroid therapies were allowed to be continued per institutional guidelines.
Patients randomized to the BAT arm were allowed to cross over to the Jakavi arm after the Day 28 visit. Tapering of Jakavi was allowed after the Day 56 visit for patients with treatment response.
Baseline demographics and disease characteristics were balanced between the two treatment arms. The median age was 54 years (range 12 to 73 years). The study included 2.9% adolescent, 59.2% male and 68.9% white patients. The majority of enrolled patients had malignant underlying disease.
The severity of acute GvHD was Grade II in 34% and 34%, Grade III in 46% and 47%, and Grade IV in 20% and 19% of the Jakavi and BAT arms, respectively.
The reasons for patients' insufficient response to corticosteroids in the Jakavi and BAT arm were i) failure in achieving a response after 7 days of corticosteroid treatment (46.8% and 40.6%, respectively), ii) failure of corticosteroid taper (30.5% and 31.6%, respectively) or iii) disease progression after 3 days of treatment (22.7% and 27.7%, respectively).
Among all patients, the most common organs involved in GvHD were skin (54.0%) and lower GI tract (68.3%). More patients in the Jakavi arm had acute GvHD involving skin (60.4%) and liver (23.4%), compared to the BAT arm (skin: 47.7% and liver: 16.1%).
The most frequently used prior systemic acute GvHD therapies were corticosteroids+CNIs (49.4% in the Jakavi arm and 49% in the BAT arm).
The primary endpoint was the overall response rate (ORR) on Day 28, defined as the proportion of patients in each arm with a complete response (CR) or a partial response (PR) without the requirement of additional systemic therapies for an earlier progression, mixed response or non-response based on investigator assessment following the criteria by Harris et al (2016).
The key secondary endpoint was the proportion of patients who achieved an ORR at Day 28 and maintained it at Day 56.
A further secondary endpoint was Failure Free Survival (FFS), a composite time to event endpoint defined as the time from randomization to i) relapse or recurrence of underlying disease, ii) non-relapse mortality, or iii) addition or initiation of another systemic therapy.
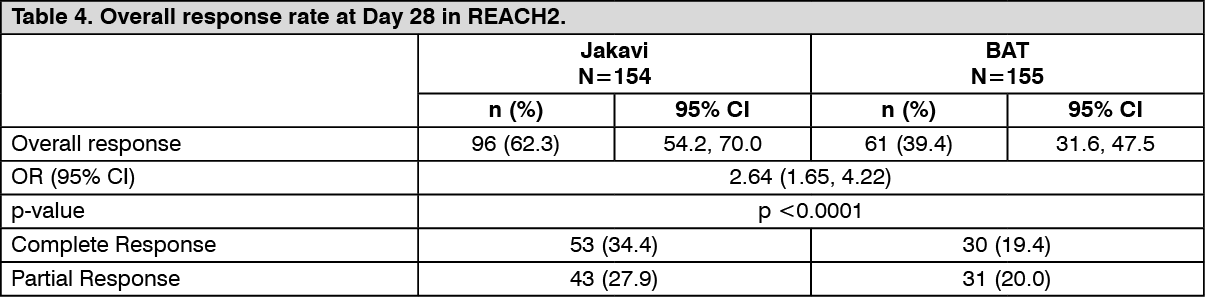
REACH2 met its primary objective. ORR at Day 28 of treatment was higher in the Jakavi arm (62.3%) compared to the BAT arm (39.4%). There was a statistically significant difference between the treatment arms (stratified Cochrane-Mantel-Haenszel test p<0.0001, one-sided, OR: 2.64; 95% CI: 1.65, 4.22).
There was also a higher proportion of complete responders in the Jakavi arm (34.4%) compared to BAT arm (19.4%).
Day-28 ORR was 76% for Grade II GvHD, 56% for Grade III GvHD, and 53% for Grade IV GvHD in the Jakavi arm, and 51% for Grade II GvHD, 38% for Grade III GvHD, and 23% for Grade IV GvHD in the BAT arm.
Among the non-responders at Day 28 in the Jakavi and BAT arms, 2.6% and 8.4% had disease progression, respectively.
Overall results are presented in Table 4. (See Table 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageThe study met its key secondary endpoint. Durable ORR at Day 56 was 39.6% (95% CI: 31.8, 47.8) in the Jakavi arm and 21.9% (95% CI: 15.7, 29.3) in the BAT arm. There was a statistically significant difference between the two treatment arms (OR: 2.38; 95% CI: 1.43, 3.94; p=0.0005). The proportion of patients with a CR was 26.6% in the Jakavi arm vs. 16.1% in the BAT arm. Overall, 49 patients originally randomized to the BAT arm crossed over to the Jakavi arm.
For the secondary endpoint FFS there were fewer events in the Jakavi arm (91; 59.1%) than in the BAT arm (121; 78.1%). Among the randomized patients, the estimated incidence rate of a FFS event at one month was lower in the Jakavi arm (18.47%; 95% CI: 12.74, 25.04) than in the BAT arm (49.13%; 95% CI: 40.94, 56.80). Additional follow-up data remain in favor of Jakavi. Median FFS with Jakavi was statistically significantly longer than with BAT (4.86 months vs. 1.02 months; HR: 0.49, 95% CI: 0.37, 0.63; p<0.0001).
Chronic graft-versus-host disease: In REACH3, 329 patients with moderate or severe corticosteroid-refractory, chronic GvHD were randomized 1:1 to Jakavi or BAT. Patients were stratified by severity of chronic GvHD at the time of randomization. Corticosteroid refractoriness was determined when patients had lack of response or disease progression after 7 days, or had disease persistence for 4 weeks or failed corticosteroid taper twice.
BAT was selected by the investigator on a patient-by-patient basis and included extracorporeal photopheresis (ECP), low dose methotrexate (MTX), mycophenolate mofetil (MMF), mTOR inhibitors (everolimus or sirolimus), infliximab, rituximab, pentostatin, imatinib, or ibrutinib.
In addition to Jakavi or BAT, patients could have received standard allogeneic stem cell transplantation supportive care including anti-infective medications and transfusion support as well as standard chronic GvHD prophylaxis and treatment medications initiated before randomization including systemic corticosteroids and CNIs (cyclosporine or tacrolimus). Topical or inhaled corticosteroid therapy was allowed to be continued per institutional guidelines.
Patients randomized to the BAT arm were allowed to cross over to the Jakavi arm after the Cycle 7 Day 1 visit (week 24). Tapering of Jakavi was allowed after the Cycle 7 Day 1 visit.
Baseline demographics and disease characteristics were balanced between the two treatment arms. The median age was 49 years (range 12 to 76 years). The study included 3.6% adolescent, 61.1% male and 75.4% white patients. The majority of enrolled patients had malignant underlying disease.
The severity at diagnosis of corticosteroid-refractory chronic GvHD was balanced between the two treatment arms, with 41% and 45% moderate, and 59% and 55% severe, in the Jakavi and the BAT arms, respectively.
Patients' insufficient response to corticosteroids in the Jakavi and BAT arm were characterized by i) a lack of response or disease progression after corticosteroid treatment for at least 7 days at 1 mg/kg/day of prednisone equivalents (37.6% and 44.5%, respectively), ii) disease persistence after 4 weeks at 0.5 mg/kg/day (35.2% and 25.6%), or iii) corticosteroid dependency (27.3% and 29.9%, respectively).
Among all patients, 73% and 45% had skin and lung involvement in the Jakavi arm, compared to 69% and 41% in the BAT arm.
The most frequently used prior systemic chronic GvHD therapies were corticosteroids only (43% in the Jakavi arm and 49% in the BAT arm) and corticosteroids+CNIs (41% patients in the Jakavi arm and 42% in the BAT arm).
The primary endpoint was the ORR on Day 1 of Cycle 7, defined as the proportion of patients in each arm with a CR or a PR without the requirement of additional systemic therapies for an earlier progression, mixed response or non-response based on investigator assessment per NIH criteria.
Key secondary endpoints were Failure Free Survival (FFS) and proportion of patients with improvement of the modified Lee symptoms score (mLSS) at Cycle 7 Day 1. FFS, a composite time to event endpoint, incorporated the earliest of the following events: i) relapse or recurrence of underlying disease or death due to underlying disease, ii) non-relapse mortality, or iii) addition or initiation of another systemic therapy for chronic GvHD.
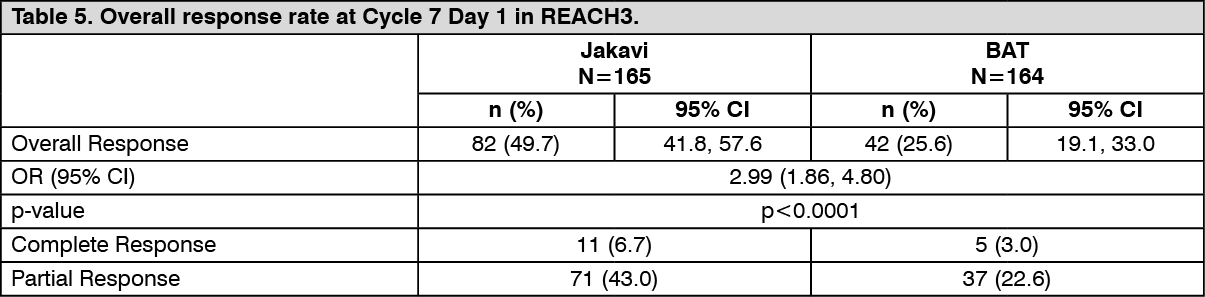
REACH3 met its primary objective. ORR at week 24 was higher in the Jakavi arm (49.7%) compared to the BAT arm (25.6%). There was a statistically significant difference between the treatment arms (stratified Cochrane-Mantel-Haenszel test p<0.0001, one-sided, OR: 2.99; 95% CI: 1.86, 4.80). Results are presented in Table 5.
Among the non-responders at Cycle 7 Day 1 in the Jakavi and BAT arms, 2.4% and 12.8% had disease progression, respectively. (See Table 5.)
 Click on icon to see table/diagram/image
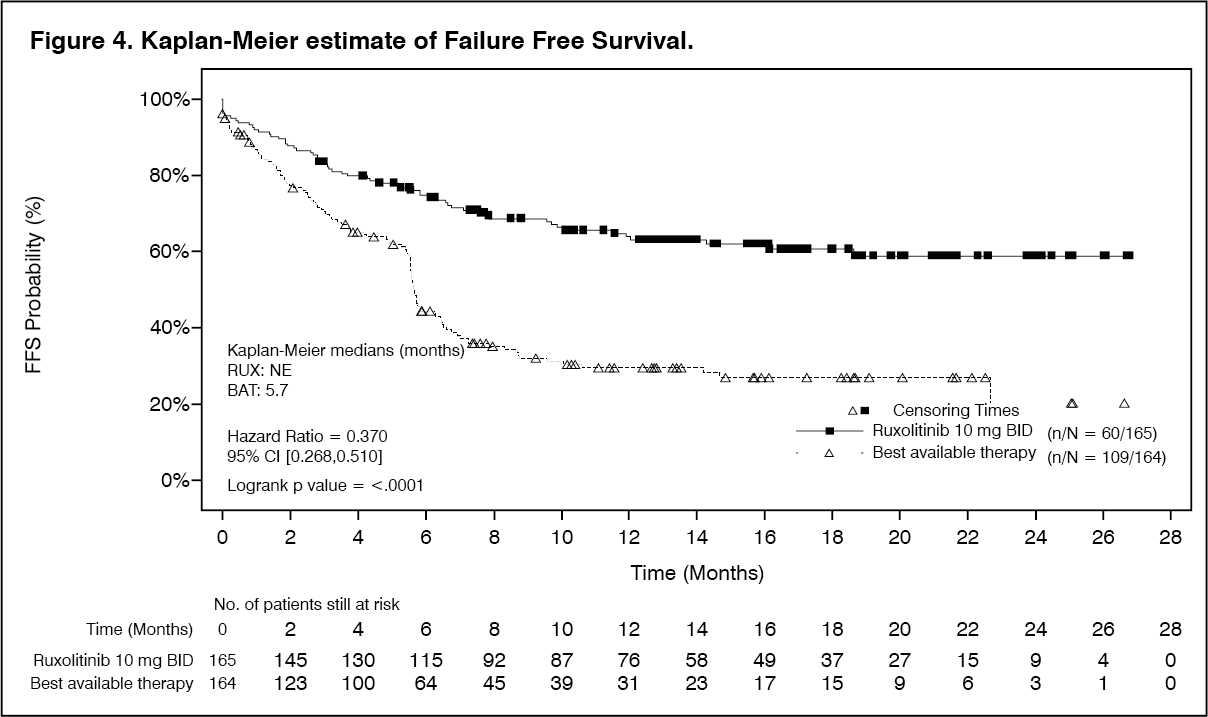
Click on icon to see table/diagram/imageBoth key secondary endpoints were also met. FFS demonstrated a statistically significant superiority of Jakavi versus BAT (HR: 0.370; 95% CI: 0.268, 0.510) with a 63% decreased risk (see Figure 4). The 6-months FFS probability (95% CI) was 74.9% (67.5%, 80.9%) and 44.5% (36.5%, 52.1%) for the Jakavi and BAT arms, respectively. The majority of FFS events were 'addition or initiation of another systemic therapy for cGvHD'. The 6-months probability for this event was 13.5% and 48.5% for the Jakavi and BAT arms, respectively. The rate of responders as per improvement of ≥7 points of total symptom score (TSS) from baseline of the mLSS showed a statistically significant difference (p=0.0011) between the Jakavi (24.2%) and BAT arms (9.8%).
Another secondary endpoint was best overall response (BOR) defined as the proportion of patients who achieved ORR (CR+PR) at any time point up to Cycle 7 Day 1. The BOR up to Cycle 7 Day 1 was higher in the Jakavi arm (76.4%) than in the BAT arm (60.4%).
The estimated probability of maintaining BOR at 12 months was higher in the Jakavi arm compared to the BAT arm (64.5% [95% CI: 58.9, 76.3] vs 40.3% [95% CI: 30.3, 50.2]). (See Figure 4.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imagePharmacokinetics: Absorption: Ruxolitinib is a Class 1 molecule under the Biopharmaceutical Classification System, with high permeability, high solubility and rapid dissolution characteristics. In clinical studies, ruxolitinib is rapidly absorbed after oral administration with maximal plasma concentration (Cmax) achieved approximately 1 hour post-dose. Based on a mass balance study in humans, oral absorption of ruxolitinib was 95% or greater. Mean ruxolitinib Cmax and total exposure (AUC) increased proportionally over a single dose range of 5 to 200 mg. There was no clinically relevant change in the PK of ruxolitinib upon administration with a high-fat meal. The mean Cmax was moderately decreased (24%) while the mean AUC was nearly unchanged (4% increase) upon dosing with a high-fat meal.
Distribution: The mean volume of distribution at steady-state is 72 L in MF patients with an inter-subject variability of 29.4% and 75 L in PV patients with an associated inter-subject variability of 22.6%. At clinically relevant concentrations of ruxolitinib, binding to plasma proteins in vitro is approximately 97%, mostly to albumin. A whole body autoradiography study in rats has shown that ruxolitinib does not penetrate the blood-brain barrier.
Biotransformation/metabolism: In vitro studies indicate that CYP3A4 and CYP2C9 are the major enzymes responsible for metabolism of ruxolitinib. Parent compound is the predominant entity in humans representing approximately 60% of the drug-related material in circulation. Two major and active metabolites were identified in plasma of healthy subjects representing 25% and 11% of parent AUC. These metabolites have one half to one fifth of the parent JAK-related pharmacological activity. The sum total of all active metabolites contribute to 18% of the overall pharmacodynamics of ruxolitinib. At clinically relevant concentrations, ruxolitinib does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 or CYP3A4 and is not a potent inducer of CYP1A2, CYP2B6 or CYP3A4 based on in vitro studies.
Elimination: Following a single oral dose of [14C]-labeled ruxolitinib in healthy adult subjects, elimination was predominately through metabolism with 74% of radioactivity excreted in urine and 22% excretion via feces. Unchanged drug accounted for less than 1% of the excreted total radioactivity. The mean elimination half-life of ruxolitinib is approximately 3 hours.
Linearity/non-linearity: Dose proportionality was demonstrated in the single and multiple dose studies.
Special populations: Effects of age, gender, or race: Based on studies in healthy subjects, no relevant differences in ruxolitinib PK were observed with regard to gender and race. In a population PK evaluation in MF patients, no relationship was apparent between oral clearance and patient age or race. Clearance was 17.7 L/h in women and 22.1 L/h in men, with 39% inter-subject variability in MF patients. Clearance was 12.7 L/h in PV patients, with a 42% inter-subject variability, and no relationship was apparent between oral clearance and gender, patient age or race in this patient population.
Pediatric: The safety and effectiveness of Jakavi in pediatric patients have not been established.
Renal impairment: Following a single Jakavi dose of 25 mg, the pharmacokinetics were similar in subjects with various degrees of renal impairment and in those with normal renal function. However, plasma AUC values of ruxolitinib metabolites tended to increase with increasing severity of renal impairment, and most markedly in the subjects with ESRD requiring hemodialysis. Ruxolitinib is not removed by dialysis. A dose modification is recommended for patients with severe renal impairment (Clcr less than 30 mL/min). For patients with ESRD a modification of the dosing schedule is recommended (see Dosage & Administration).
Hepatic impairment: Following a single Jakavi dose of 25 mg in patients with varying degrees of hepatic impairment, the pharmacokinetics and pharmacodynamics of ruxolitinib were assessed. The mean AUC for ruxolitinib was increased in patients with mild, moderate and severe hepatic impairment by 87%, 28% and 65%, respectively, compared to patients with normal hepatic function and indicating no clear relationship to the degree of hepatic impairment based on Child-Pugh scores. The terminal elimination half-life was prolonged in patients with hepatic impairment compared to healthy controls (4.1-5.0 hours versus 2.8 hours). A dose reduction is recommended for MF and PV patients with hepatic impairment (see Dosage & Administration).
Mild, moderate or severe hepatic impairment in patients with GvHD was not found to have a significant impact on any parameter in the Population PK model.
Toxicology: Non-Clinical Safety Data: Ruxolitinib has been evaluated in safety pharmacology, repeat dose toxicity, genotoxicity, reproductive toxicity studies and a carcinogenicity study. Target organs associated with the pharmacological action of ruxolitinib in repeat dose studies include bone marrow, peripheral blood and lymphoid tissues. Infections generally associated with immunosuppression were noted in dogs. Adverse decreases in blood pressure along with increases in heart rate were noted in a dog telemetry study, and an adverse decrease in minute volume was noted in a respiratory study in rats. The margins (based on unbound Cmax) at the non-adverse effect level in the dog and rat studies were 15.7-fold and 10.4-fold greater, respectively, than the maximum human recommended 25 mg twice daily dose. No effects were noted in an evaluation of the neuropharmacologic effects of ruxolitinib.
Administration of ruxolitinib to juvenile rats resulted in effects on growth and bone measures. Ruxolitinib was administered daily by oral gavage at doses from 1.5 to 75 mg/kg/day from days 7 (the human equivalent of a newborn) to 63 post-partum (pp), 15 mg/kg/day from days 14 (the human equivalent of 1 year of age) to 63 pp and 5, 15 and 60 mg/kg/day from days 21 (the human equivalent of 2 to 3 years of age) to 63 pp. Doses ≥30 mg/kg/day (1,200 ng*h/mL based on unbound AUC) resulted in fractures and early termination of the groups when treatment started on day 7 pp. Reduced bone growth was observed at doses ≥5 mg/kg/day (≥150 ng*h/mL based on unbound AUC) when treatment started on day 7 pp and at ≥15 mg/kg/day (≥150 ng*h/mL based on unbound AUC) when treatment started on day 14 pp or day 21 pp. Based on unbound AUC, fractures and reduced bone growth occurred at exposures 13- and 1.5- fold the exposure in adult patients at the maximum recommended dose of 25 mg b.i.d, respectively. The effects were generally more severe when administration was initiated earlier in the postnatal period. Other than the effects on bone development, the toxicity profile in juvenile rats was comparable to that observed in adult rats.
Reproductive toxicity data are quoted in Females and males of reproductive potential under Use in Pregnancy & Lactation. Ruxolitinib was not mutagenic or clastogenic. Ruxolitinib was not carcinogenic in the Tg.rasH2 transgenic mouse model nor in a 2-year study in rats.