Therapeutic/Pharmacologic Class of Drug: Antibody drug conjugate antineoplastic agent.
ATC Code: L01XC14.
Pharmacology: Pharmacodynamics: Mechanism of Action: Kadcyla, trastuzumab emtansine, is a HER2-targeted antibody-drug conjugate which contains the humanized anti-HER2 IgG1, trastuzumab, covalently linked to the microtubule inhibitory drug DM1 (a maytansine derivative) via the stable thioether linker MCC (4-[N-maleimidomethyl] cyclohexane-1-carboxylate). Emtansine refers to the MCC-DM1 complex. An average of 3.5 DM1 molecules are conjugated to each molecule of trastuzumab.
Conjugation of DM1 to trastuzumab confers selectivity of the cytotoxic agent for HER2-overexpressing tumor cells, thereby increasing intracellular delivery of DM1 directly to malignant cells. Upon binding to HER2, trastuzumab emtansine undergoes receptor-mediated internalization and subsequent lysosomal degradation, resulting in release of DM1-containing cytotoxic catabolites (primarily lysine-MCC-DM1).
Kadcyla has the mechanisms of action of both trastuzumab and DM1: Trastuzumab emtansine, like trastuzumab, binds to domain IV of the HER2 extracellular domain (ECD), as well as to Fcγ receptors and complement C1q. In addition, Kadcyla, like trastuzumab, inhibits shedding of the HER2 ECD, inhibits signaling through the phosphatidylinositol 3-kinase (PI3-K) pathway, and mediates antibody-dependent cell-mediated cytotoxicity (ADCC) in human breast cancer cells that overexpress HER2.
DM1, the cytotoxic drug component of Kadcyla, binds to tubulin. By inhibiting tubulin polymerization, both DM1 and Kadcyla cause cells to arrest in the G2/M phase of the cell cycle, ultimately leading to apoptotic cell death. Results from
in vitro cytotoxicity assays show that DM1 is 20-200 times more potent than taxanes and vinca alkaloids.
The MCC linker is designed to limit systemic release and increase targeted delivery of DM1, as demonstrated by detection of very low levels of free DM1 in plasma.
Clinical/Efficacy Studies: Efficacy:
Early Breast Cancer: KATHERINE (BO27938) was a randomized, multicenter, open-label trial of 1486 patients with HER2-positive, early breast cancer with residual invasive tumor in the breast and/or axillary lymph nodes following taxane and trastuzumab-based therapy as part of a neoadjuvant regimen before trial enrollment. Patients received radiotherapy and/or hormonal therapy concurrent with study treatment as per local guidelines. Breast tumor samples were required to show HER2 overexpression defined as 3+ IHC or ISH amplification ratio ≥ 2.0 determined at a central laboratory. Patients were randomized (1:1) to receive trastuzumab or KADCYLA. Randomization was stratified by clinical stage at presentation, hormone receptor status, preoperative HER2-directed therapy (trastuzumab, trastuzumab plus additional HER2-directed agent[s]), and pathological nodal status evaluated after preoperative therapy.
KADCYLA was given intravenously at 3.6 mg/kg on Day 1 of a 21-day cycle. Trastuzumab was given intravenously at 6 mg/kg on Day 1 of a 21-day cycle. Patients were treated with KADCYLA or trastuzumab for a total of 14 cycles unless there was recurrence of disease, withdrawal of consent, or unacceptable toxicity, whichever occurred first. At the time of the primary analysis, median treatment duration was 10 months (range: 1-12) for KADCYLA, and median treatment duration 10 months (range: 1-13) for trastuzumab. Patients who discontinued KADCYLA could complete the duration of their intended study treatment up to 14 cycles of HER2-directed therapy with trastuzumab, if appropriate, based on toxicity considerations and investigator discretion.
The primary efficacy endpoint of the study was Invasive Disease Free Survival (IDFS). IDFS was defined as the time from the date of randomization to first occurrence of ipsilateral invasive breast tumor recurrence, ipsilateral local or regional invasive breast cancer recurrence, distant recurrence, contralateral invasive breast cancer, or death from any cause. Additional endpoints included IDFS including second primary non-breast cancer, disease free survival (DFS), overall survival (OS), and distant recurrence-free interval (DRFI).
Patient demographics and baseline tumor characteristics were balanced between treatment arms. The median age was approximately 49 years (range 23-80 years), 72.8% were White, 8.7% were Asian and 2.7% were Black or African American. All but 5 patients were women. 22.5 percent of patients were enrolled in North America, 54.2% in Europe and 23.3% throughout the rest of the world. Tumor prognostic characteristics including hormone receptor status (positive: 72.3%, negative: 27.7%), clinical stage at presentation (inoperable: 25.3%, operable: 74.8%) and pathological nodal status after preoperative therapy (node positive: 46.4%, node negative not evaluated: 53.6%) were similar in the study arms.
The majority of the patients (76.9%) had received an anthracycline-containing neoadjuvant chemotherapy regimen. 19.5% of patients received another HER2-targeted agent in addition to trastuzumab as a component of neoadjuvant therapy. Pertuzumab was the second therapy in 93.8% of patients who received a second neoadjuvant HER2-directed agent.
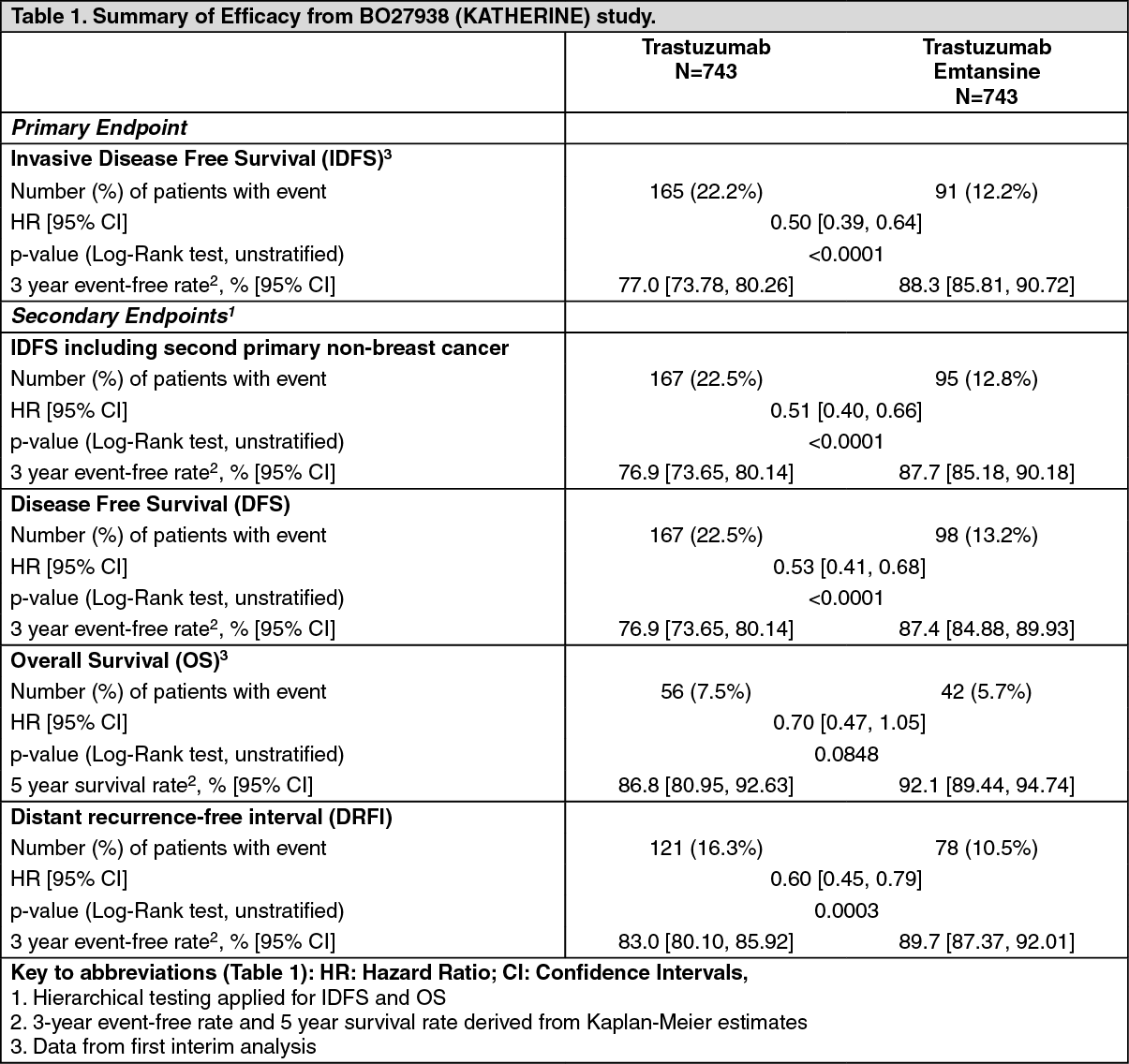
A clinically meaningful and statistically significant improvement in IDFS was observed in patients who received trastuzumab emtansine compared with trastuzumab (HR = 0.50, 95% CI [0.39, 0.64], p <0.0001), corresponding to a 50% reduction in risk of an IDFS event. Estimates of 3 years IDFS rates were 88.3% vs. 77.0% in trastuzumab emtansine vs. trastuzumab arms, respectively. See Table 1 and Figures 1 and 2.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In KATHERINE, consistent treatment benefit of Kadcyla for IDFS was seen in all the pre specified subgroups evaluated, supporting the robustness of the overall result (see Figure 3). In the subgroup of patients with hormone receptor-negative disease (n=412, 27.7%), the hazard ratio for IDFS was 0.50 (95% CI: 0.33, 0.74). In the subgroup of patients with hormone receptor-positive disease (n=1074, 72.3%), the hazard ratio for IDFS was 0.48 (95% CI: 0.35, 0.67). In the subgroup of patients who received neoadjuvant trastuzumab with chemotherapy (n=1196, 80.5%), the hazard ratio for IDFS was 0.49 (95% CI: 0.37, 0.65). In the subgroup of patients who received neoadjuvant trastuzumab plus a second HER2- directed therapy with chemotherapy (n=290, 19.5%), the hazard ratio for IDFS was 0.54 (95% CI: 0.27, 1.06). Patients who received Perjeta as a second neoadjuvant HER2-directed agent (n=272, 93.8%), had an IDFS hazard ratio of 0.50 (95% CI: 0.25, 1.00). The IDFS hazard ratio in patients who were node-positive after preoperative therapy (n=689, 46.4%) was 0.52 (95% CI: 0.38, 0.71). In patients who were node-negative or not evaluated after preoperative therapy (n=797, 53.6%), the hazard ratio for IDFS was 0.44 (95% CI: 0.28, 0.68). (See Figure 3.)
Click on icon to see table/diagram/image
Patient reported outcomes included the assessment of patient-reported global health status, role and physical function, and treatment symptoms using the EORTC QLQ-C30 and EORTC QLQ-BR23. In the analyses of patient-reported outcomes, a 10-point difference was considered clinically meaningful.
Patients' function, global health status, and symptom scores showed no clinically meaningful change from baseline over the course of treatment or during follow-up. Mean change from baseline at Cycle 11 for physical function was -0.6 (95% CI -1.9-0.7) in the trastuzumab emtansine arm and 1.8 (95% 0.6-3.1) in the trastuzumab arm; global health status was 0.4 (95% CI -2.2-1.3) in the trastuzumab emtansine arm and 1.4 (95% CI -0.2-3.0) in the trastuzumab arm. No clear differences were observed in function, global health status, or symptoms between the two treatment arms and at no timepoint during the study were the average scores on those scales of patients in the trastuzumab emtansine arm clinically worse than those of patients in the trastuzumab arm.
Metastatic Breast Cancer: A Phase III, randomized, multicenter, international, open-label clinical trial (TDM4370g/BO21977) was conducted in patients with HER2-positive unresectable locally advanced breast cancer or MBC who had received prior taxane and trastuzumab-based therapy, including patients who received prior therapy with trastuzumab and a taxane in the adjuvant setting and who relapsed within six months of completing adjuvant therapy. Prior to enrollment, breast tumor samples were required to be centrally confirmed for HER2-positive disease defined as a score of 3+ by IHC or gene amplification by ISH. Baseline patient and tumor characteristics were well balanced between treatment groups. For patients randomized to Kadcyla, the median age was 53 years, most patients were female (99.8%), the majority were Caucasian (72%), and 57% had estrogen-receptor and/or progesterone-receptor positive disease. The study compared the safety and efficacy of Kadcyla with that of lapatinib plus capecitabine. A total of 991 patients were randomized with Kadcyla or lapatinib plus capecitabine as follows: Kadcyla Arm: Kadcyla 3.6 mg/kg intravenously (IV) over 30-90 minutes on Day 1 of a 21-day cycle.
Control Arm (lapatinib plus capecitabine): lapatinib 1250 mg/day orally once per day of a 21-day cycle plus capecitabine 1000 mg/m
2 orally twice daily on Days 1-14 of a 21-day cycle.
The co-primary efficacy endpoints of the study were progression-free survival (PFS) as assessed by an independent review committee (IRC), and overall survival (OS) and landmark (1-year and 2-year) survival rates.
Time to symptom progression, as defined by a 5-point decrease in the score derived from the trials outcome index-breast (TOI-B) subscale of the Functional Assessment of Cancer Therapy-Breast Quality of Life (FACT-B QoL) questionnaire was also assessed during the clinical trial. A change of 5 points in the TOI-B is considered clinically significant. (See Table 2.)
Click on icon to see table/diagram/image
A treatment benefit was seen in the subgroup of patients who had not received any prior systemic anti-cancer therapy in the metastatic setting (n=118); hazard ratios for PFS and OS were 0.51 (95% CI: 0.30, 0.85) and 0.61 (95% CI: 0.32, 1.16), respectively. The median PFS and OS for the KADCYLA group were 10.8 months and not reached, respectively, compared with 5.7 months and 27.9 months, respectively, for the lapatinib plus capecitabine group. (See Figures 4 and 5.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
A randomized, multicenter, open-label phase II, study (TDM4450g/BO21976) evaluated the effects of Kadcyla versus trastuzumab plus docetaxel in patients with HER2-positive MBC who had not received prior chemotherapy for metastatic disease. Patients were randomized to receive Kadcyla 3.6 mg/kg IV every 3 weeks (n=67) or trastuzumab 8 mg/kg IV loading dose followed by 6 mg/kg IV every 3 weeks plus docetaxel 75-100 mg/m
2 IV every 3 weeks (n=70).
The primary endpoint was PFS assessed by investigator. The median PFS was 9.2 months in the trastuzumab plus docetaxel arm and 14.2 months in the Kadcyla arm (hazard ratio, 0.59; p = 0.035), with a median follow-up of approximately 14 months in both arms. The ORR was 58.0% with trastuzumab plus docetaxel and 64.2% with Kadcyla. The median duration of response was not reached with Kadcyla vs. median duration 9.5 months in the control arm.
The worsening of the FACT-B TOI scores was delayed in the Kadcyla arm compared with the control arm (median time to symptom progression was 7.5 months in the Kadcyla arm vs. 3.5 months in the control arm; hazard ratio, 0.58; p = 0.022).
A Phase II, single-arm, open-label study (TDM4374g) evaluated the effects of Kadcyla in patients with HER2-positive incurable locally advanced, or MBC. All patients were previously treated with HER2-directed therapies (trastuzumab and lapatinib), and chemotherapy (anthracycline, taxane, and capecitabine) in the neoadjuvant, adjuvant, locally advanced, or metastatic setting. The median number of anti-cancer agents that patients received in any setting was 8.5 (range, 5-19) and in the metastatic setting was 7.0 (range, 3-17), including all agents intended for the treatment of breast cancer.
Patients (n=110) received 3.6 mg/kg of Kadcyla intravenously every 3 weeks until disease progression or unacceptable toxicity.
The key efficacy analyses were ORR based on independent radiologic review and duration of objective response. The ORR was 32.7% (95% CI: 24.1, 42.1), n=36 responders, by both IRC and investigator review. The median duration of response by IRC was not reached (95% CI, 4.6 months to not estimable).
Immunogenicity: As with all therapeutic proteins, there is the potential for an immune response to Kadcyla. A total of 1243 patients from seven clinical studies were tested at multiple time points for anti-drug antibody (ADA) responses to Kadcyla. Following Kadcyla dosing, 5.1% (63/1243) of patients tested positive for anti-Kadcyla antibodies at one or more post-dose time points. In the Phase I and Phase II studies, 6.4% (24/376) of patients tested positive for anti-Kadcyla antibodies. In the EMILIA study (TDM4370g/BO21977), 5.2% (24/466) of patients tested positive for anti-Kadcyla antibodies, of which 13 were also positive for neutralizing antibodies. In the KATHERINE (BO27938) study, 3.7% (15/401) of patients tested positive for anti-Kadcyla antibodies, of which 5 of were also positive for neutralizing antibodies. Due to the low incidence of ADA, conclusions cannot be made on the impact of anti-Kadcyla antibodies on the pharmacokinetics, safety, and efficacy of Kadcyla.
Immunogenicity assay results are highly dependent on several factors including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications and underlying disease. For these reasons, comparison of incidence of antibodies to Kadcyla with the incidence of antibodies to other products may be misleading.
Pharmacokinetics: The population pharmacokinetic analysis of trastuzumab emtansine suggested no difference in Kadcyla exposure based on disease status (adjuvant vs. metastatic setting).
Absorption: Kadcyla is administered IV. There have been no studies performed with other routes of administration.
Distribution: Kadcyla when administered intravenously every 3 weeks exhibited linear pharmacokinetics across doses ranging from 2.4 to 4.8 mg/kg; patients who received doses less than or equal to 1.2 mg/kg had faster clearance.
Patients in Study TDM4370g/BO21977 and Study BO29738 who received 3.6 mg/kg of Kadcyla intravenously every 3 weeks had a mean maximum serum concentration (Cmax) of trastuzumab emtansine in Cycle 1 of 83.4 (± 16.5) μg/mL and 72.6 (± 24.3) μg/mL, respectively. Based on population pharmacokinetic analysis, following intravenous administration, the central volume of distribution of trastuzumab emtansine was (3.13 L) and approximated that of plasma volume.
Metabolism: Kadcyla is expected to undergo catabolism by means of proteolysis in cellular lysosomes, with no significant involvement of cytochrome P450 isoenzymes. Catabolites including Lys-MCC-DM1, MCC-DM1 and DM1 are detected at low levels in human plasma. In Study TDM4370g/BO21977 and Study BO29738, mean maximum DM1 levels in Cycle 1 following Kadcyla administration were consistently low and averaged 4.61 (± 1.61) ng/mL and 4.71 (± 2.25) ng/mL, respectively.
In vitro metabolism studies in human liver microsomes suggest that DM1, a component of trastuzumab emtansine, is metabolized mainly by CYP3A4 and to a lesser extent by CYP3A5.
Elimination: Based on population pharmacokinetic (PK) analysis, following IV administration of Kadcyla in patients with HER2-positive metastatic breast cancer, the clearance of trastuzumab emtansine was 0.68 L/day and the elimination half-life (t
1/2) was approximately 4 days. No accumulation of trastuzumab emtansine was observed after repeated dosing of IV infusion every 3 weeks.
Based on a population PK analysis (n=671), body weight, albumin, sum of longest diameter of target lesions by RECIST, HER2 ECD, baseline trastuzumab concentrations and AST were identified as statistically significant covariates for trastuzumab emtansine pharmacokinetic parameters. However, the magnitude of effect of these covariates on trastuzumab emtansine exposure suggests that, with the exception of body weight, these covariates are unlikely to have any clinically meaningful effect on trastuzumab emtansine exposure. Therefore, the body weight based dose of 3.6 mg/kg every 3 weeks without correction for other covariates is considered appropriate. In nonclinical studies, catabolites of trastuzumab emtansine including DM1, Lys-MCC-DM1, and MCC-DM1 are mainly excreted in the bile with minimal elimination in urine.
Pharmacokinetics in Special Populations: The population pharmacokinetic analysis of trastuzumab emtansine showed that race did not appear to influence the pharmacokinetics of Kadcyla. Because most of the patients in Kadcyla clinical studies were females, effect of gender on the pharmacokinetics of Kadcyla was not formally evaluated.
Geriatric Population: The population pharmacokinetic analysis of trastuzumab emtansine showed that age did not affect the pharmacokinetics of Kadcyla. No significant difference was observed in the pharmacokinetics of trastuzumab emtansine among patients <65 years (n=577), patients between 65-75 years (n=78) and patients >75 years (n=16).
Renal impairment: The population pharmacokinetic analysis of trastuzumab emtansine showed that creatinine clearance does not affect pharmacokinetics of Kadcyla. Pharmacokinetics of trastuzumab emtansine in patients with mild (creatinine clearance CLcr 60-89 mL/min, n=254) or moderate (CLcr 30 to 59 mL/min, n=53) renal impairment were similar to those in patients with normal renal function (CLcr ≥90 mL/min, n=361). Pharmacokinetic data in patients with severe renal impairment (CLcr 15-29 mL/min) is limited (n=1), therefore no dosage recommendations can be made.
Hepatic impairment: The liver is a primary organ for eliminating DM1 and DM1-containing catabolites. The pharmacokinetics of trastuzumab emtansine and DM1-containing catabolites were evaluated after the administration of 3.6 mg/kg of Kadcyla to metastatic HER2-positive breast cancer patients with normal hepatic function (n=10), mild (Child-Pugh A; n=10) and moderate (Child-Pugh B; n=8) hepatic impairment.
Plasma concentrations of DM1 and DM1-containing catabolites (Lys-MCC-DM1 and MCC-DM1) were low and comparable between patients with and without hepatic impairment.
Systemic exposures (AUC) of trastuzumab emtansine at Cycle 1 in patients with mild and moderate hepatic impairment were approximately 38% and 67% lower than that of patients with normal hepatic function, respectively. Trastuzumab emtansine exposure (AUC) at Cycle 3 after repeated dosing in patients with mild or moderate hepatic dysfunction was within the range observed in patients with normal hepatic function.
No formal pharmacokinetic study has been conducted and no population PK data was collected in patients with severe hepatic impairment (Child-Pugh class C).
Toxicology: Nonclinical Safety: Carcinogenicity: No carcinogenicity studies have been performed to establish the carcinogenic potential of Kadcyla.
Genotoxicity: No evidence of mutagenic activity was observed in an
in vitro bacterial reverse mutation assay of DM1. In an
in vivo micronucleus assay of trastuzumab emtansine in cynomolgus monkeys, no evidence of chromosomal damage to bone marrow cells was observed. However, in a rat bone marrow micronucleus assay, DM1 was positive for micronuclei formation after a single low dose in the DM1 concentration range measured in humans given trastuzumab emtansine, confirming that Kadcyla is an aneugen and/or clastogen.
Impairment of Fertility: No fertility studies in animals have been performed to evaluate the effect of Kadcyla. However, based on results from general animal toxicity studies, adverse effects on fertility can be expected.
Reproductive toxicity: Dedicated embryo-fetal development studies have not been conducted in animals with trastuzumab emtansine. Developmental toxicity of trastuzumab has been identified in the clinical setting although it was not predicted in the non-clinical program. In addition, developmental toxicity of maytansine has been identified in non-clinical studies which suggests that DM1, the microtubule-inhibiting cytotoxic maytansinoid drug component of trastuzumab emtansine, will be similarly teratogenic and potentially embryotoxic.
Other: Not applicable.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image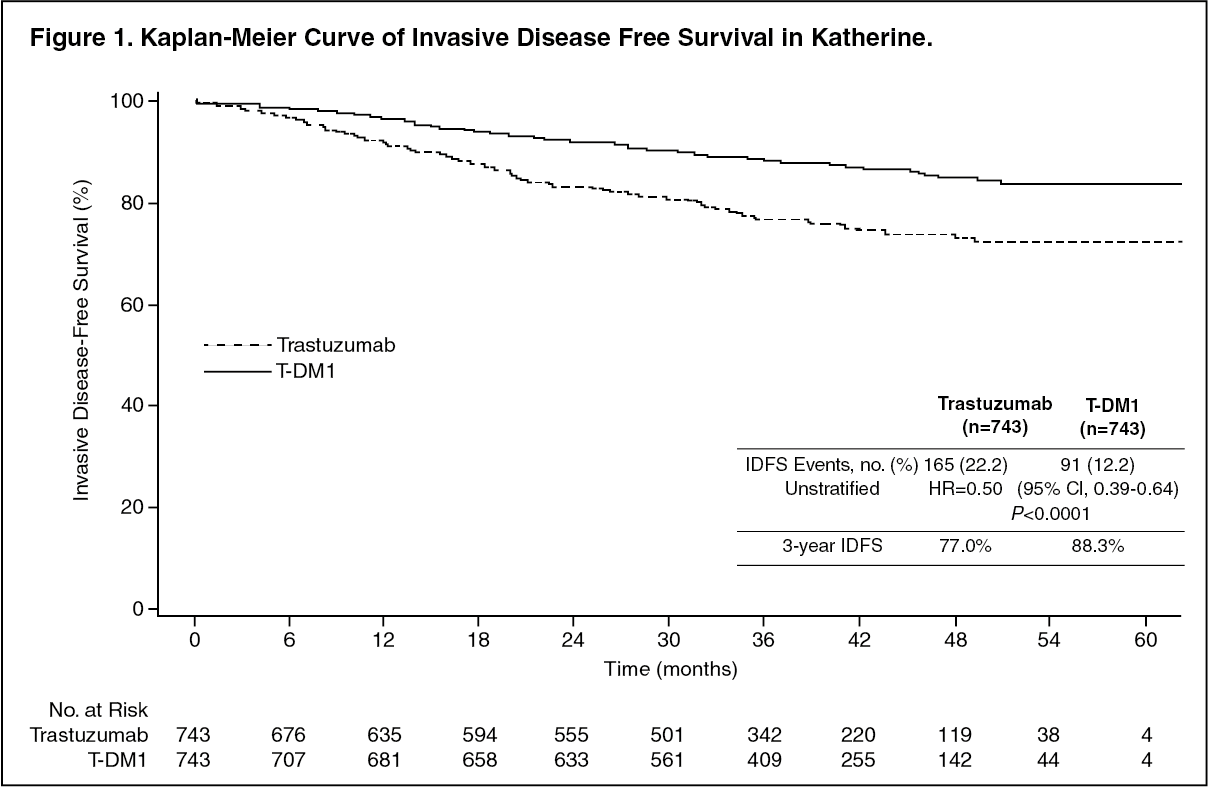 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image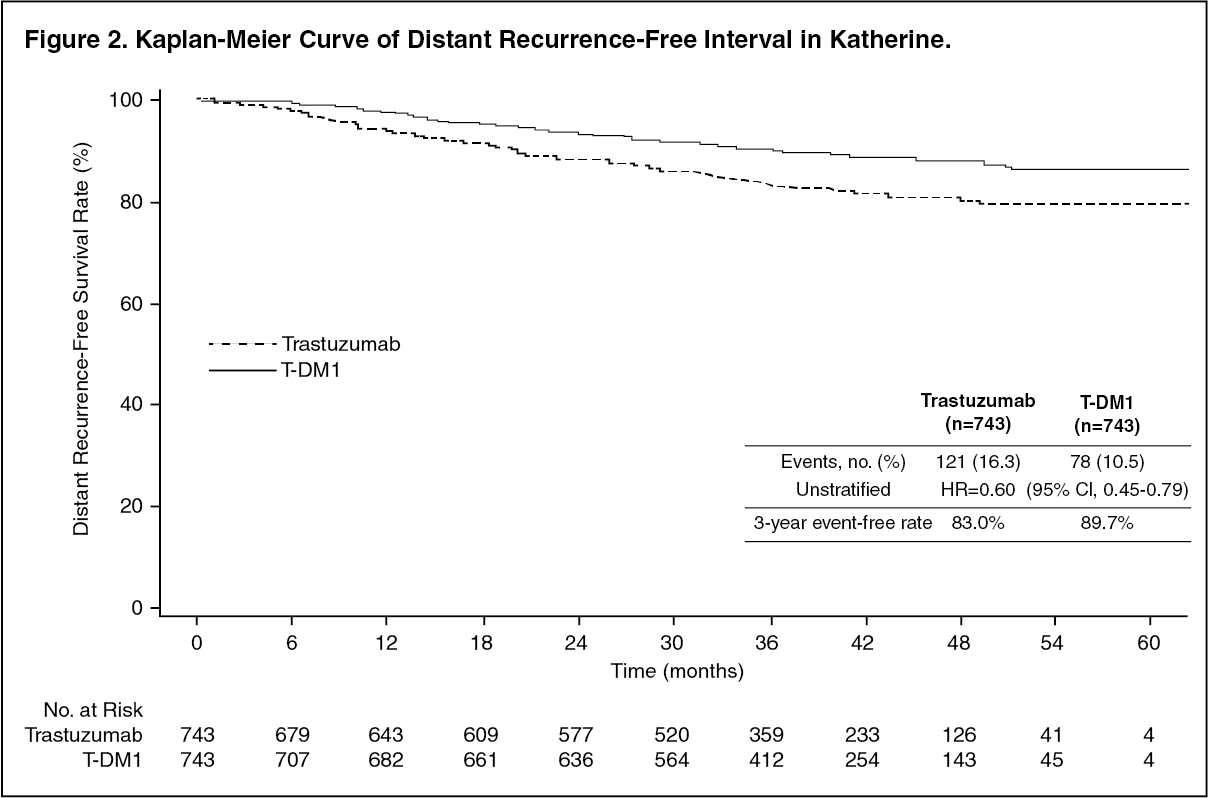 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image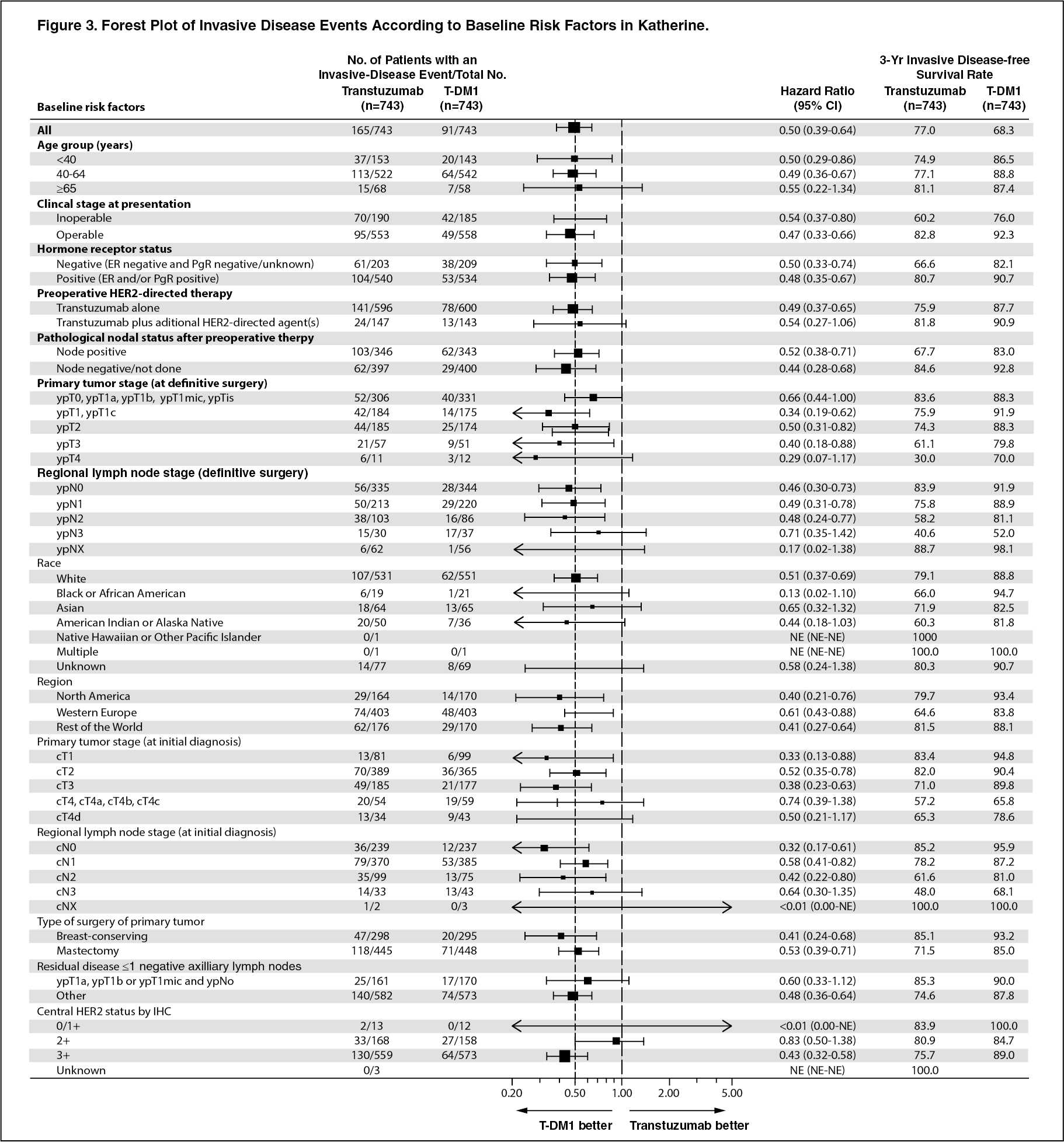 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image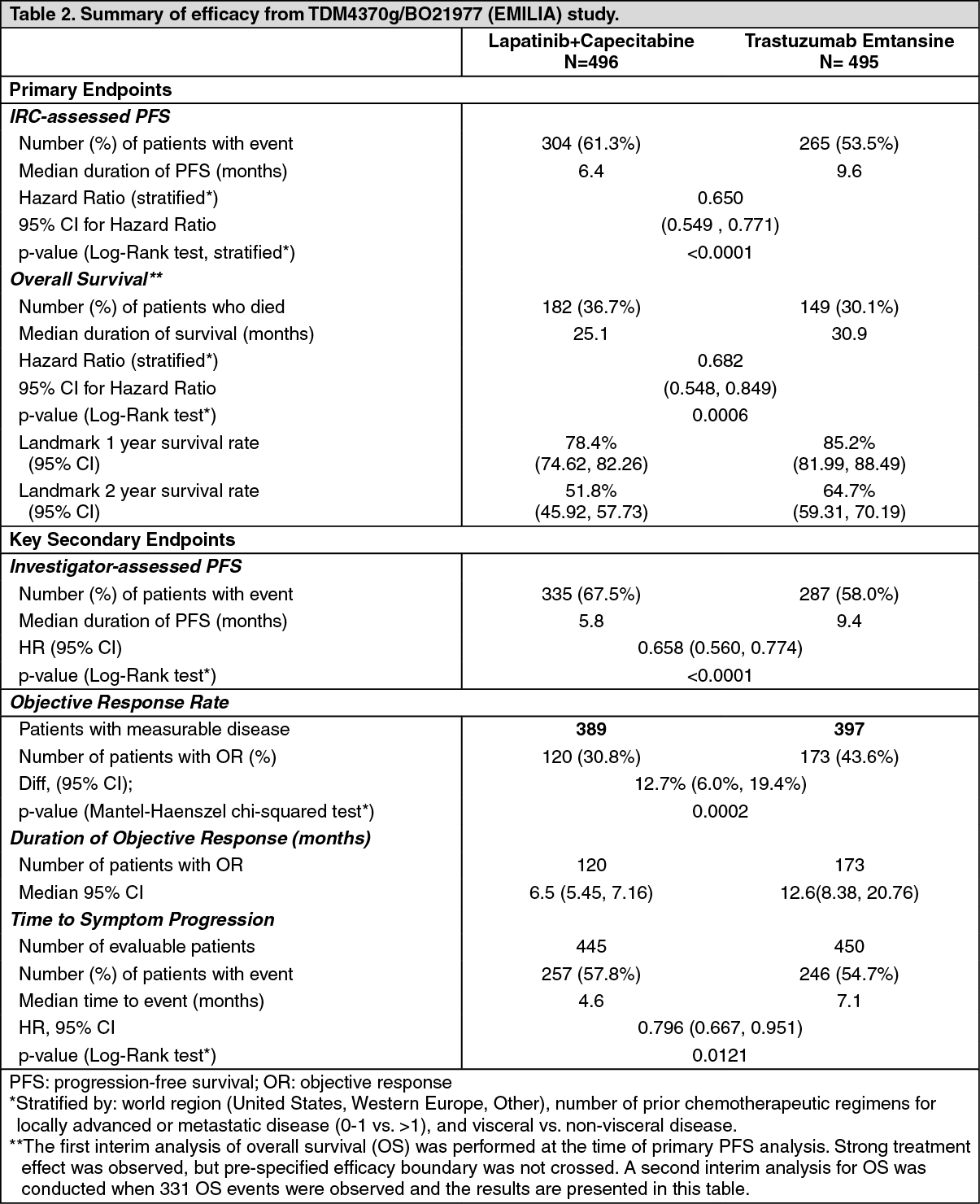 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image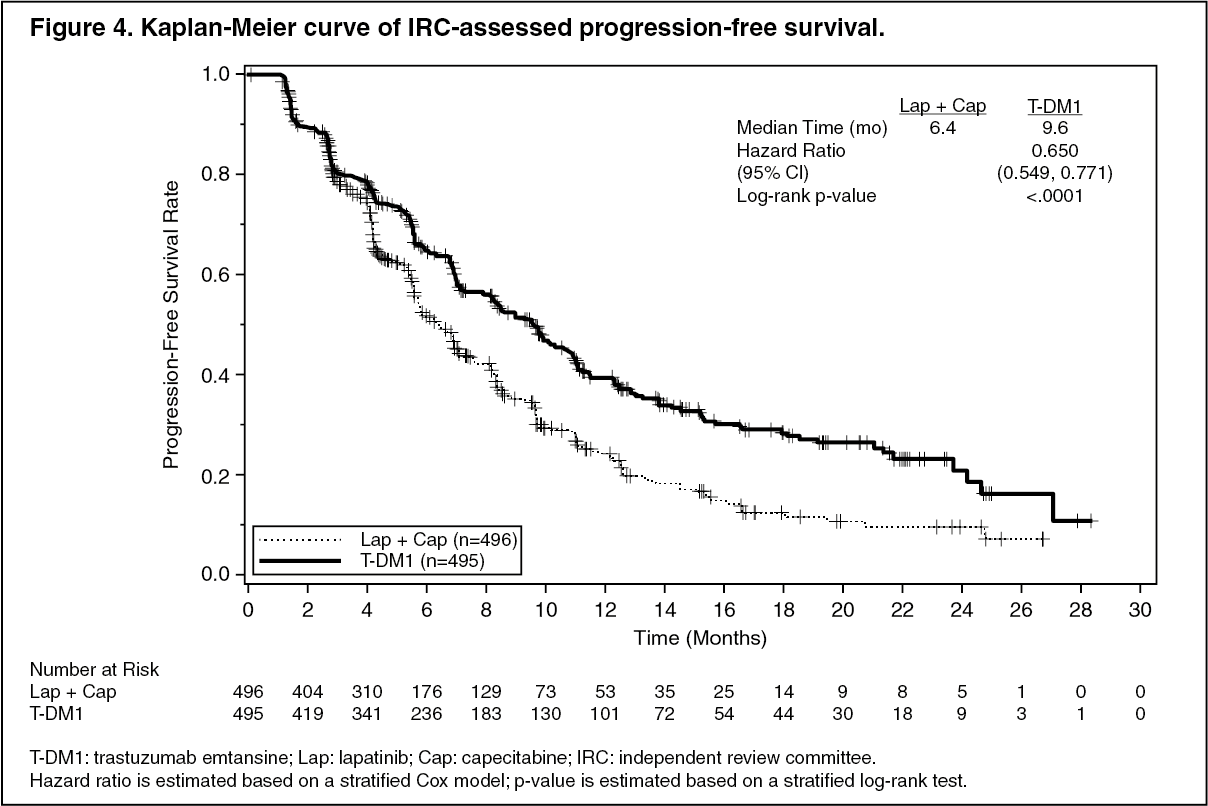 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image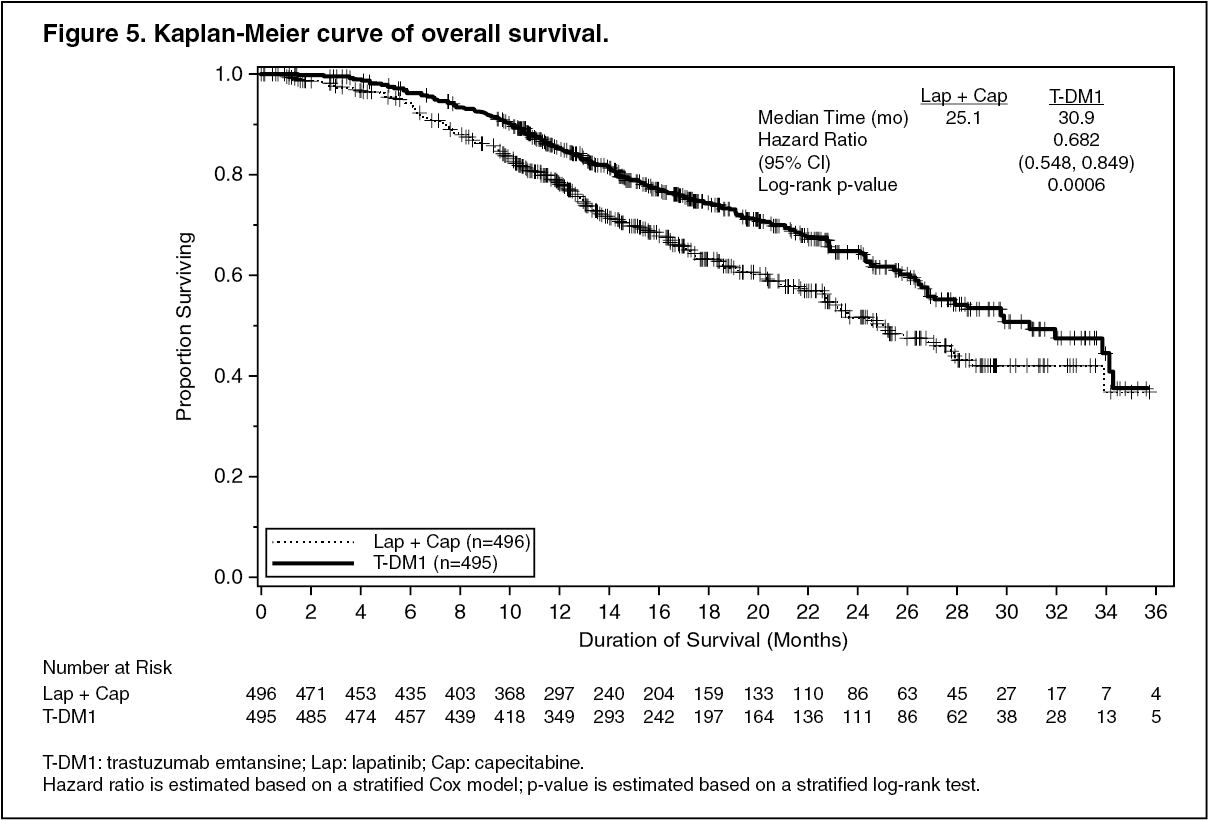 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image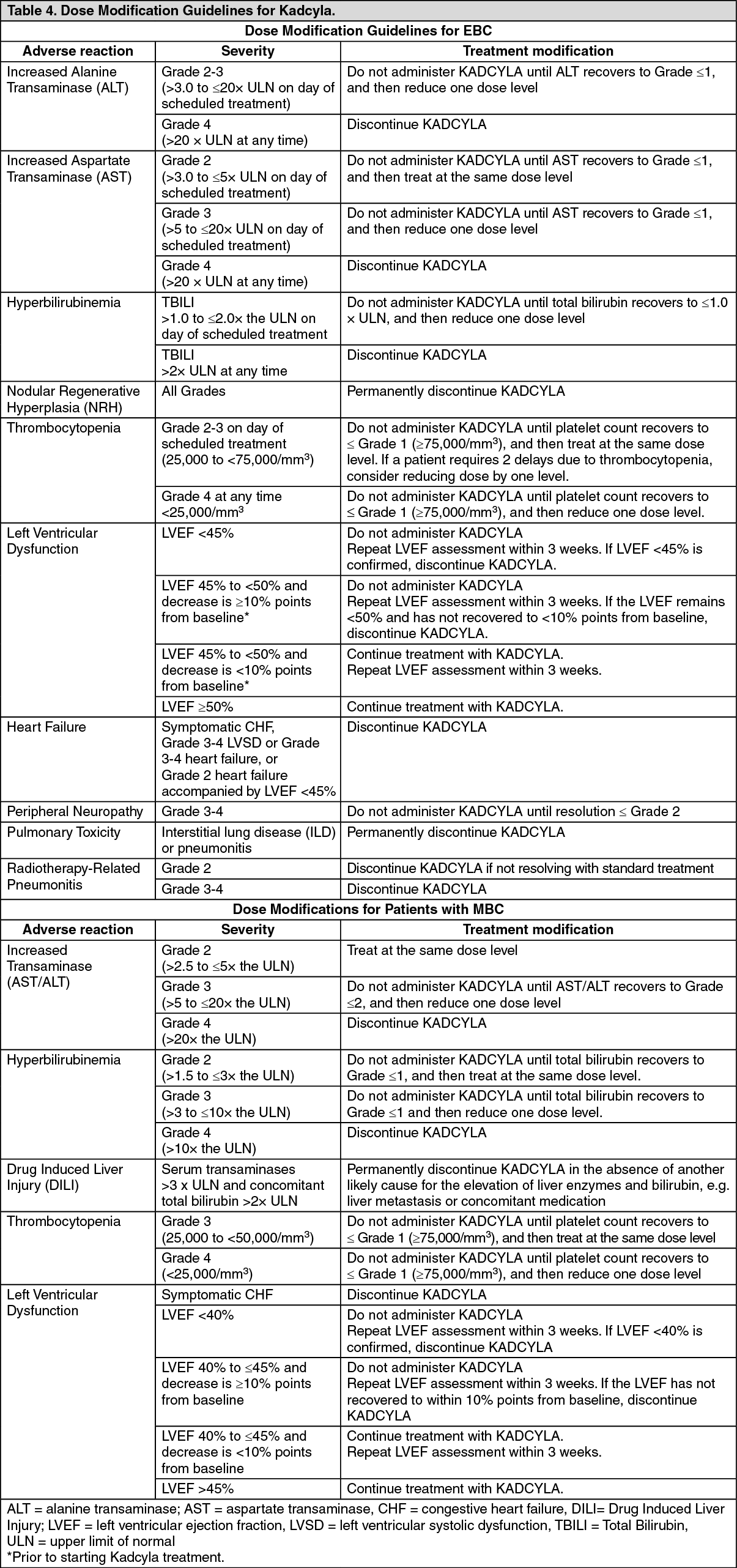 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image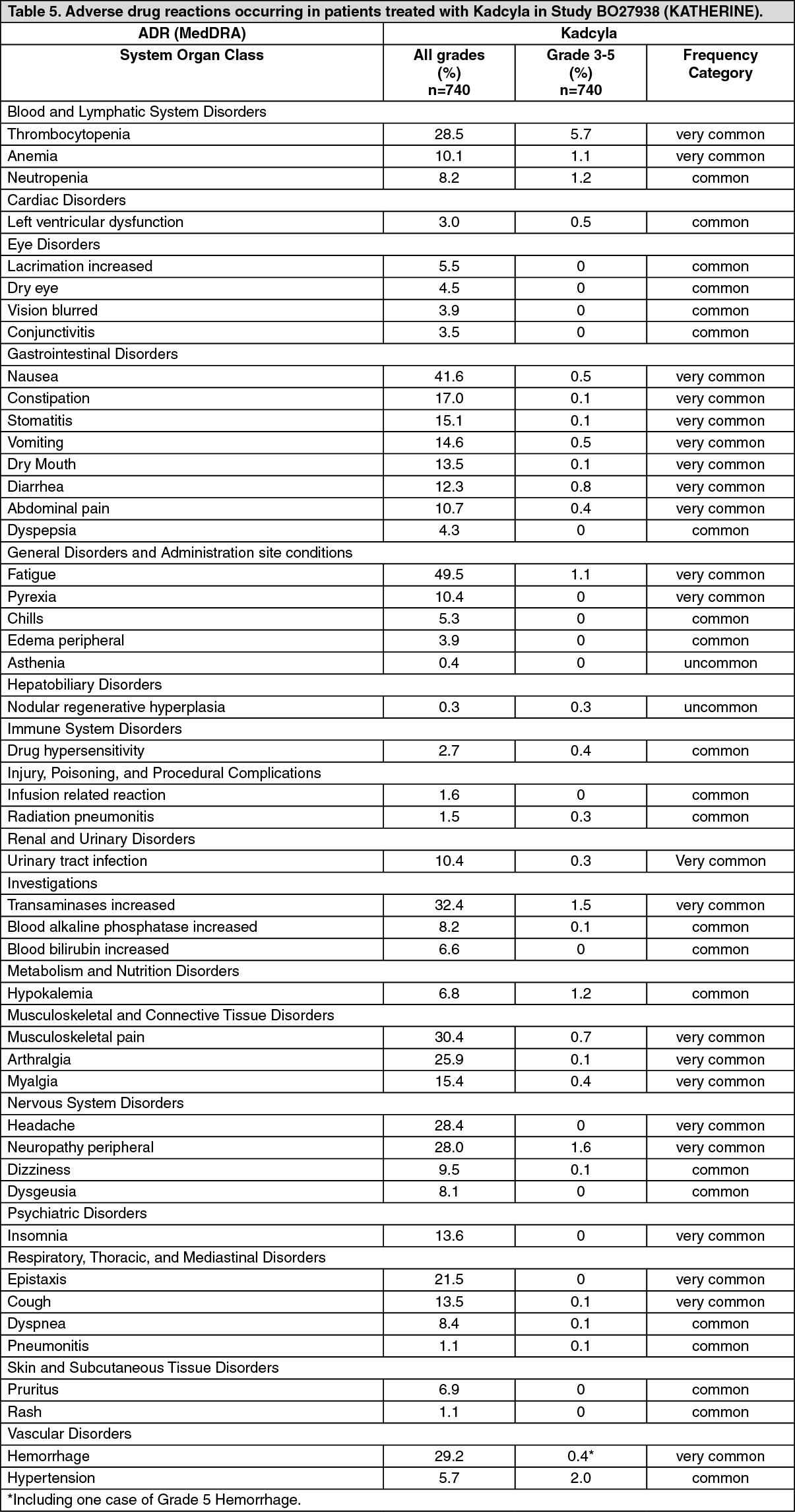 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image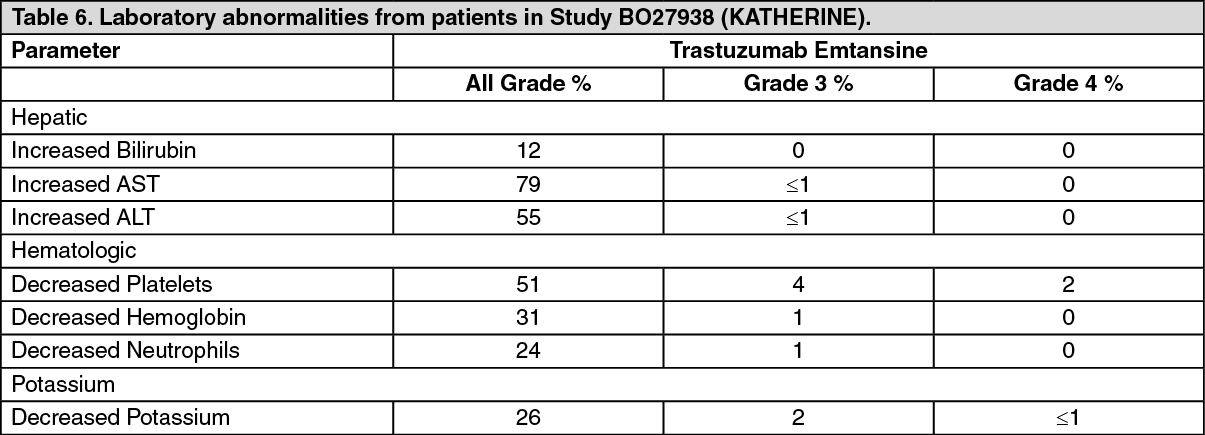 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image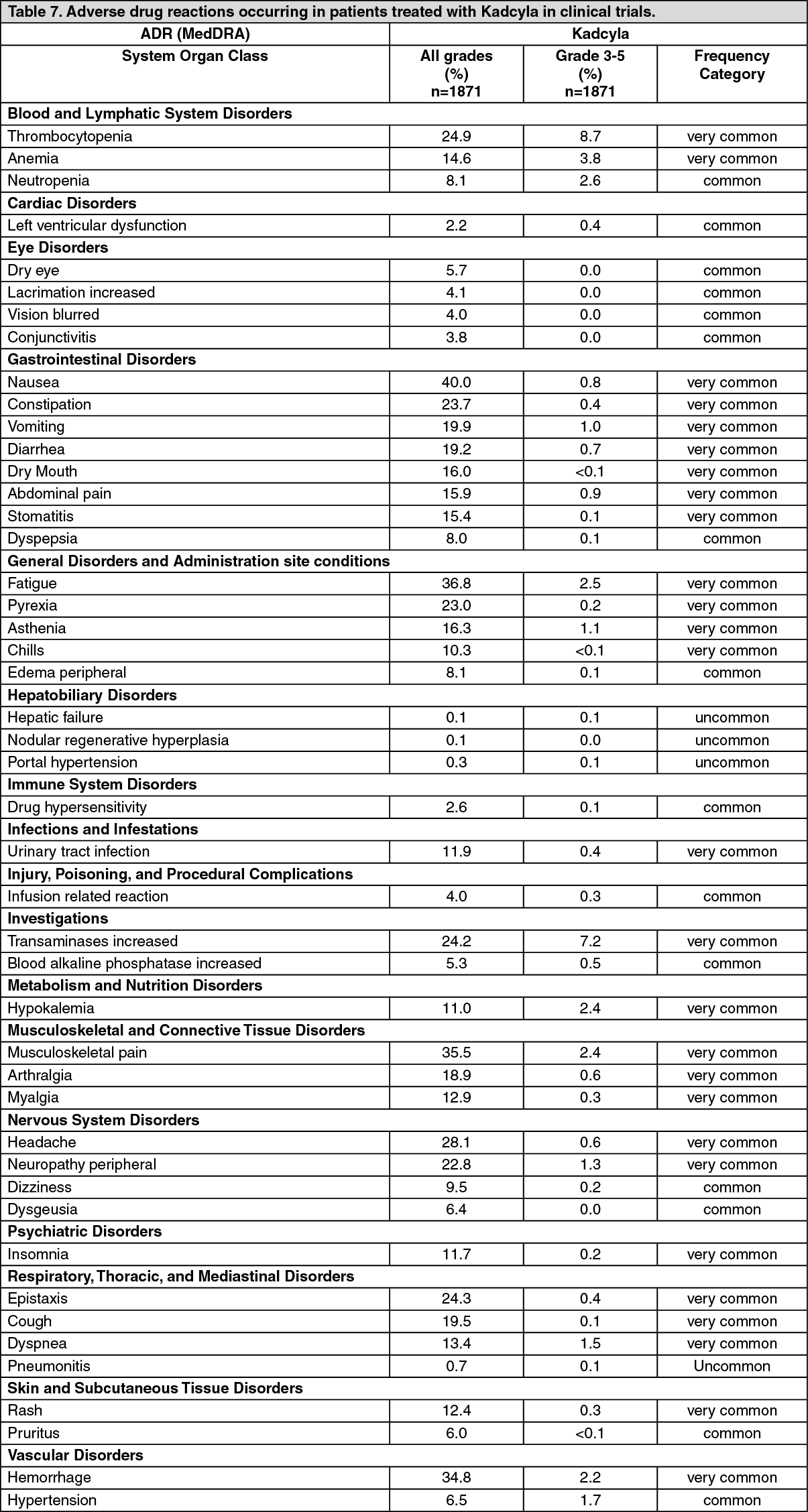 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image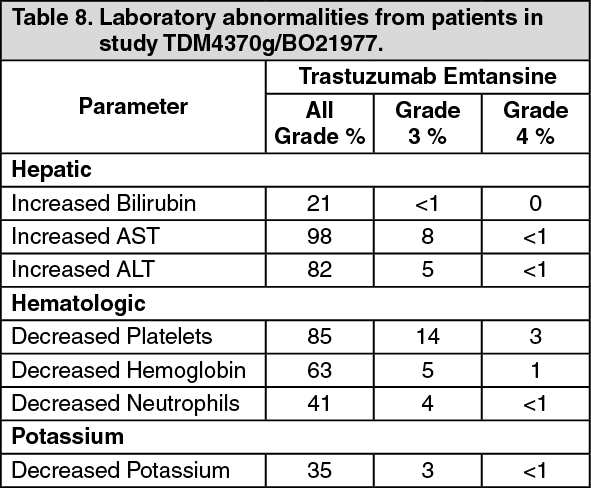 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image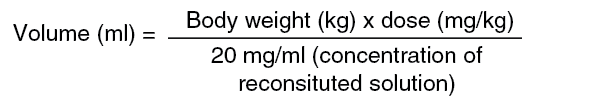 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out